PyTorch implementation of a Self-Organizing Map. The implementation makes possible the use of a GPU if available for faster computations. It follows the scikit package semantics for training and usage of the model. It also includes runnable scripts to avoid coding.
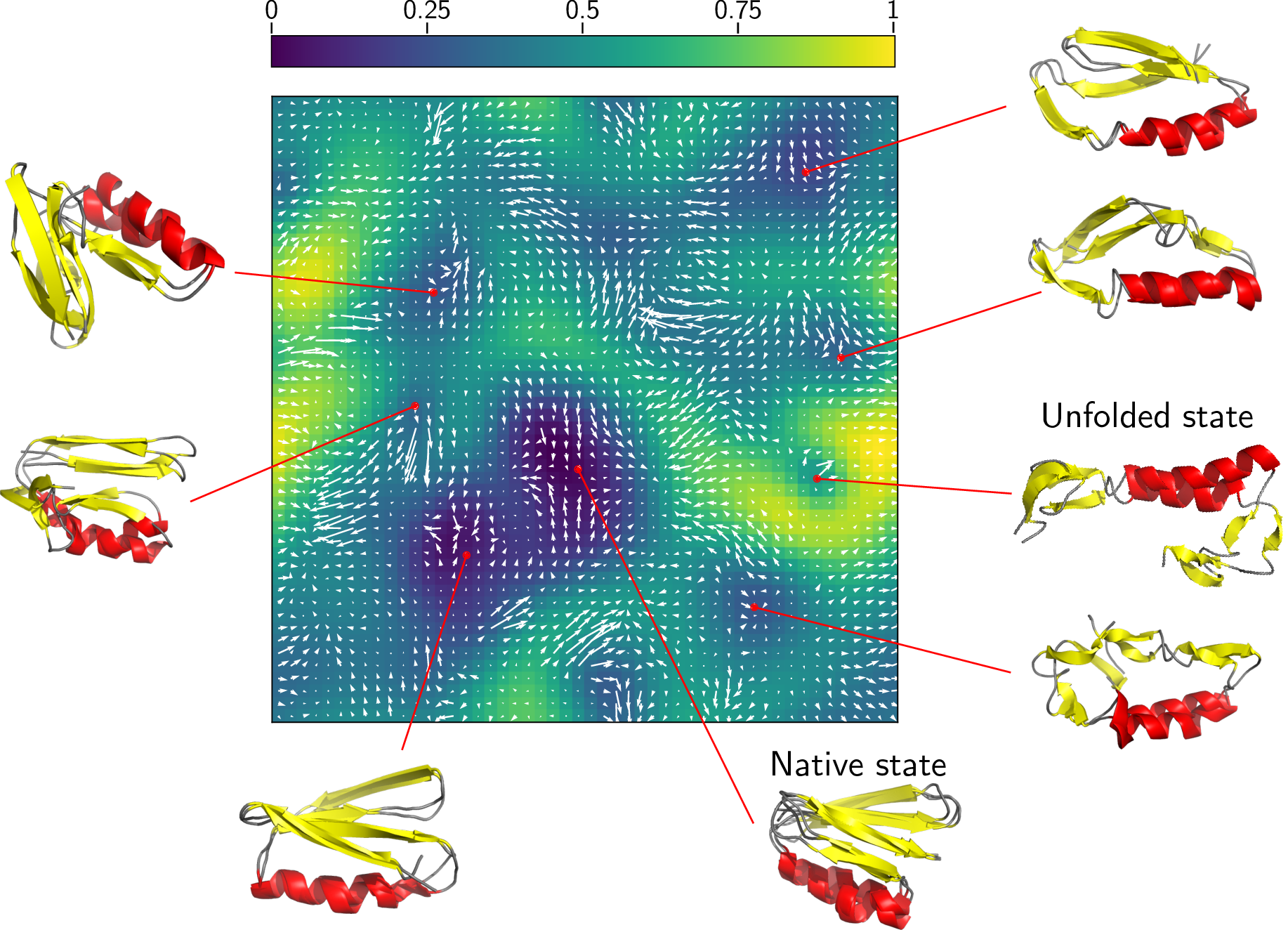
Example of a MD clustering using quicksom:
The SOM object requires PyTorch installed.
It has dependencies in numpy, scipy and scikit-learn and scikit-image. The MD application requires pymol to load the trajectory that is not included in the dependencies
To set up the project, we suggest using conda environments. Install PyTorch and run :
pip install quicksom
The SOM object can be created using any grid size, with a optional periodic topology. One can also choose optimization parameters such as the number of epochs to train or the batch size
To use it, we include three scripts to :
- fit a SOM
- to optionally build the clusters manually with a gui
- to predict cluster affectations for new data points
$ quicksom_fit -h
usage: quicksom_fit [-h] -i IN_NAME [-o OUT_NAME] [-m M] [-n N] [--norm NORM]
[--periodic] [--n_iter N_ITER] [--batch_size BATCH_SIZE]
[--alpha ALPHA] [--sigma SIGMA] [--scheduler SCHEDULER]
optional arguments:
-h, --help show this help message and exit
-i IN_NAME, --in_name IN_NAME
name of the .npy to use
-o OUT_NAME, --out_name OUT_NAME
name of pickle to dump
-m M, --m M The width of the som
-n N, --n N The height of the som
--norm NORM The p norm to use
--periodic if set, periodic topology is used
--n_iter N_ITER The number of iterations
--batch_size BATCH_SIZE
The batch size to use
--alpha ALPHA The initial learning rate
--sigma SIGMA The initial sigma for the convolution
--scheduler SCHEDULER
Which scheduler to use, can be linear, exp or half
$ quicksom_gui -h
usage: quicksom_gui [-h] [-i IN_NAME] [-o OUT_NAME]
optional arguments:
-h, --help show this help message and exit
-i IN_NAME, --in_name IN_NAME
name of the som to load
-o OUT_NAME, --out_name OUT_NAME
name of the som to dump if we want it different
$ quicksom_predict -h
usage: quicksom_predict [-h] [-i IN_NAME] [-o OUT_NAME] [-s SOM_NAME]
[--recompute_cluster] [--batch BATCH] [--subset]
All the indices are starting from 1.
optional arguments:
-h, --help show this help message and exit
-i IN_NAME, --in_name IN_NAME
name of the npy file to use
-o OUT_NAME, --out_name OUT_NAME
name of txt to dump
-s SOM_NAME, --som_name SOM_NAME
name of pickle to load
--recompute_cluster if set, periodic topology is used
--batch BATCH Batch size
--subset Use the user defined clusters instead of the expanded
partition.
The SOM object is also importable from python scripts to use directly in your analysis pipelines :
import pickle
import numpy
import torch
from quicksom.som import SOM
# Get data
device = 'cuda' if torch.cuda.is_available() else 'cpu'
X = numpy.load('contact_desc.npy')
X = torch.from_numpy(X)
X = X.float()
X = X.to(device)
# Create SOM object and train it, then dump it as a pickle object
m, n = 100, 100
dim = X.shape[1]
niter = 5
batch_size = 100
som = SOM(m, n, dim, niter=niter, device=device)
learning_error = som.fit(X, batch_size=batch_size)
som.to_device('cpu')
pickle.dump(som, open('som.pickle', 'wb'))
# Usage on the input data, predicted_clusts is an array of length n_samples with clusters affectations
som = pickle.load(open('som.pickle', 'rb'))
som.to_device(device)
predicted_clusts, errors = som.predict_cluster(X)dcd2npy: Pymolmdx: Pymol, pymol-psico
To set these dependencies up using conda, just type :
conda install -c schrodinger pymol pymol-psico
The SOM algorithm can efficiently map MD trajectories for analysis and clustering purposes.
The script dcd2npy can be used to select a subset of atoms from a trajectory in dcd format,
align it and save the selection as a npy file that can be handled by the command quicksom_fit.
$ dcd2npy -h
usage: dcd2npy [-h] --pdb PDB --dcd DCD --select SELECTION
Convert a dcd trajectory to a numpy object
optional arguments:
-h, --help show this help message and exit
--pdb PDB Topology PDB file
--dcd DCD DCD trajectory file
--select SELECTION Atoms to select
The following commands can be applied for a MD clustering.
$ dcd2npy --pdb data/2lj5.pdb --dcd data/2lj5.dcd --select 'name CA'
dcdplugin) detected standard 32-bit DCD file of native endianness
dcdplugin) CHARMM format DCD file (also NAMD 2.1 and later)
ObjectMolecule: read set 1 into state 2...
[...]
ObjectMolecule: read set 301 into state 302...
PyMOL not running, entering library mode (experimental)
Coords shape: (301, 228)
$ quicksom_fit -i data/2lj5.npy -o data/som_2lj5.p --n_iter 100 --batch_size 50 --periodic --alpha 0.5
1/100: 50/301 | alpha: 0.500000 | sigma: 25.000000 | error: 397.090729 | time 0.387760
4/100: 150/301 | alpha: 0.483333 | sigma: 24.166667 | error: 8.836357 | time 5.738029
7/100: 250/301 | alpha: 0.466667 | sigma: 23.333333 | error: 8.722509 | time 11.213565
[...]
91/100: 50/301 | alpha: 0.050000 | sigma: 2.500000 | error: 5.658005 | time 137.348755
94/100: 150/301 | alpha: 0.033333 | sigma: 1.666667 | error: 5.373021 | time 142.033695
97/100: 250/301 | alpha: 0.016667 | sigma: 0.833333 | error: 5.855451 | time 147.203326
quicksom_gui -i data/som_2lj5.pWe now have a trained SOM and we can use several functionalities such as clustering input data points and grouping them into separate dcd files, creating a dcd with one centroid per fram or plotting of the U-Matrix and its flow.
quicksom_predict -i data/2lj5.npy -o data/2lj5 -s data/som_2lj5.p
This command generates 3 files:
$ ls data/2lj5_*.txt
data/2lj5_bmus.txt
data/2lj5_clusters.txt
data/2lj5_codebook.txt
containing the data:
- Best Matching Unit with error for each data point
- Cluster assignment
- Assignment for each SOM cell of the closest data point (BMU with minimal error). -1 means no assignment
$ head -3 data/2lj5_bmus.txt
38.0000 36.0000 4.9054
37.0000 47.0000 4.6754
2.0000 27.0000 7.0854
$ head -3 data/2lj5_clusters.txt
4 9 22 27 28 32 39 43 44 45 46 48 75 77 78 92 94 98 102 119 126 127 142 147 153 154 162 171 172 180 185 189 190 191 197 206 218 223 226 227 235 245 255 265 285 286 292 299
3 5 7 10 14 21 23 26 29 33 37 51 54 55 63 64 70 74 80 82 83 84 85 86 88 99 103 104 106 107 108 116 121 123 129 131 132 133 139 140 146 148 150 155 159 161 163 165 170 173 179 181 183 200 209 214 217 220 221 228 229 231 237 239 240 241 247 248 250 251 256 258 260 267 275 277 278 279 287 291 293 296 297 301
1 2 8 11 12 13 15 17 18 19 20 24 25 30 31 35 38 41 42 50 52 56 58 60 61 62 65 66 68 69 71 72 73 79 87 89 90 91 93 95 96 97 101 105 109 110 112 113 114 118 120 122 124 125 130 134 136 137 138 141 143 144 145 151 152 156 157 158 160 166 168 169 174 175 176 177 178 184 187 188 193 195 201 205 208 210 211 212 213 215 216 222 225 230 232 233 234 236 242 244 246 249 252 253 254 259 261 262 264 266 268 270 271 272 274 276 280 282 283 284 288 289 290 295 298 300
$ quicksom_extract -h
Extract clusters from a dcd file
quicksom_extract -p pdb_file -t dcd_file -c cluster_file
quicksom_extract -p data/2lj5.pdb -t data/2lj5.dcd -c data/2lj5_clusters.txt
$ ls -v data/cluster_*.dcd
data/cluster_1.dcd
data/cluster_2.dcd
data/cluster_3.dcd
data/cluster_4.dcd
grep -v "\-1" data/2lj5_codebook.txt > _codebook.txt
mdx --top data/2lj5.pdb --traj data/2lj5.dcd --fframes _codebook.txt --out data/centroids.dcd
rm _codebook.txt
python3 -c 'import pickle
import matplotlib.pyplot as plt
som=pickle.load(open("data/som_2lj5.p", "rb"))
plt.matshow(som.umat)
plt.savefig("data/umat_2lj5.png")
'
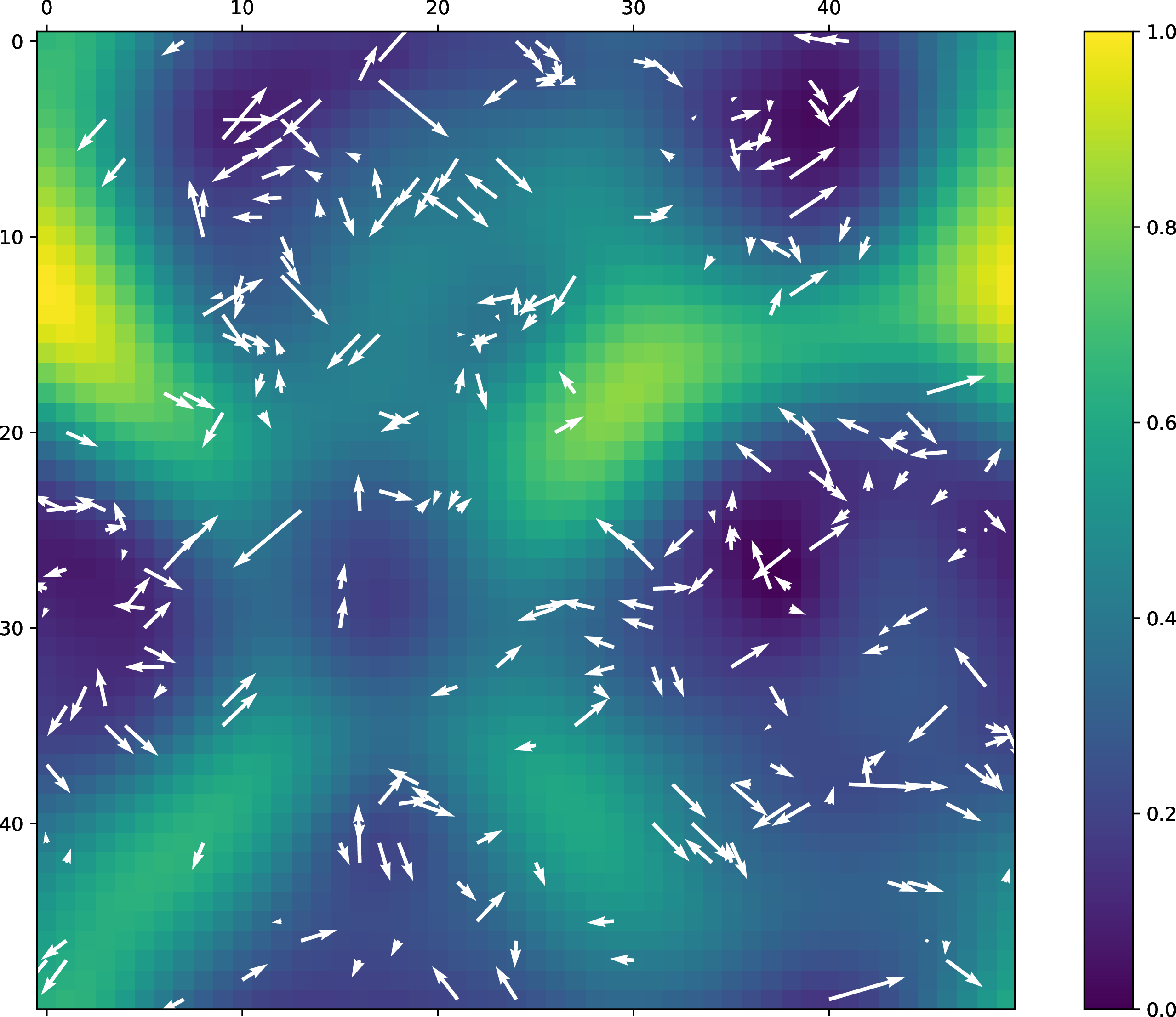
The flow of the trajectory can be projected onto the U-matrix using the following command:
$ quicksom_flow -h
usage: quicksom_flow [-h] [-s SOM_NAME] [-b BMUS] [-n] [-m] [--stride STRIDE]
Plot flow for time serie clustering.
optional arguments:
-h, --help show this help message and exit
-s SOM_NAME, --som_name SOM_NAME
name of the SOM pickle to load
-b BMUS, --bmus BMUS BMU file to plot
-n, --norm Normalize flow as unit vectors
-m, --mean Average the flow by the number of structure per SOM
cell
--stride STRIDE Stride of the vectors field
With this toy example, we get the following plot:
$ quicksom_project -h
usage: quicksom_project [-h] [-s SOM_NAME] [-b BMUS] -d DATA
Plot flow for time serie clustering.
optional arguments:
-h, --help show this help message and exit
-s SOM_NAME, --som_name SOM_NAME
name of the SOM pickle to load
-b BMUS, --bmus BMUS BMU file to plot
-d DATA, --data DATA Data file to project