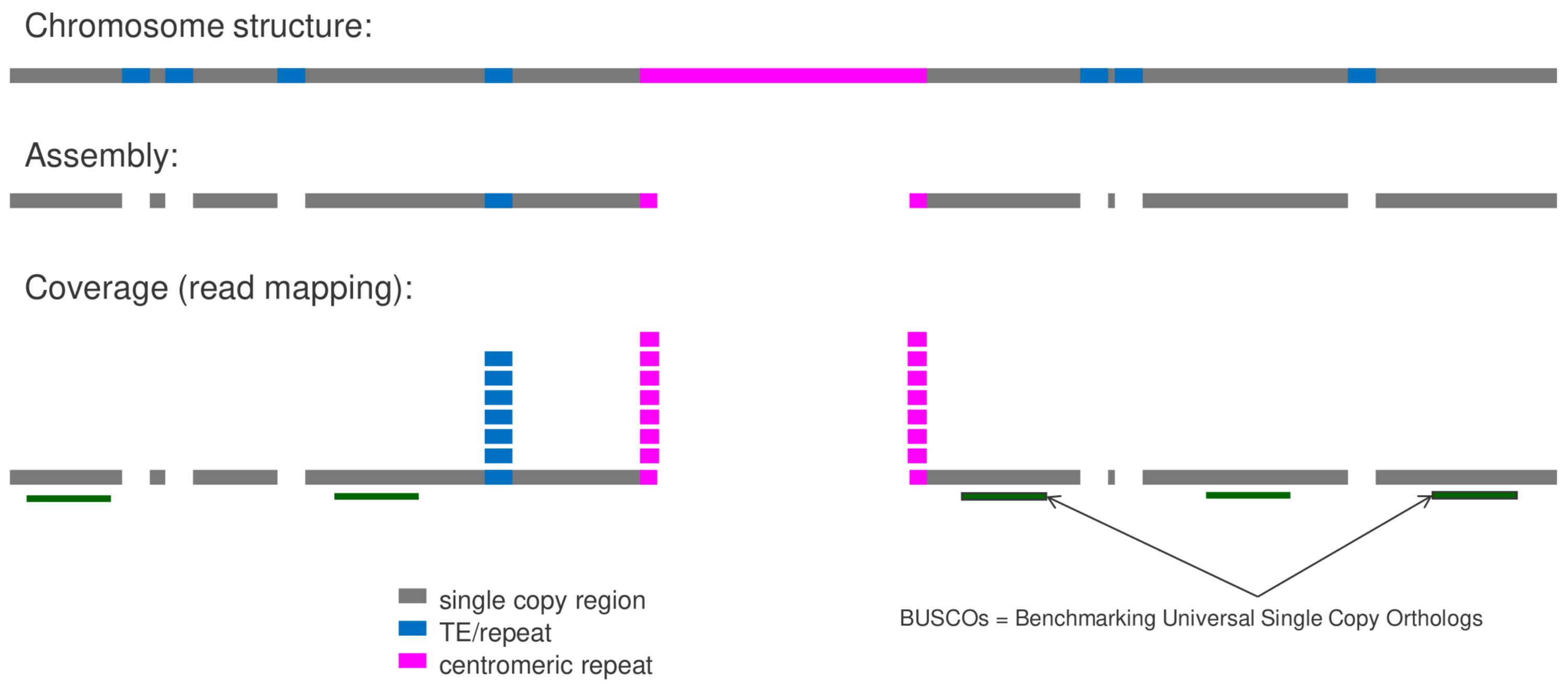
MGSE can harness the power of files generated in genome sequencing projects to predict the genome size. Required are the FASTA file containing a high continuity assembly and a BAM file with all available reads mapped to this assembly. The script construct_cov_file.py (https://doi.org/10.1186/s12864-018-5360-z) allows the generation of a COV file based on the (sorted) BAM file (also possible via MGSE directly). Next, this COV file can be used by MGSE to calculate the coverage in provided reference regions and to calculate the total number of mapped bases. Both values are subjected to the genome size estimation. Providing accurate reference regions is crucial for this genome size estimation. Different alternatives were evaluated and actual single copy BUSCOs (https://busco.ezlab.org/) appear to be the best choice. Running BUSCO prior to MGSE will generate all necessary files.
Usage:
python3 MGSE3.py [--cov <COV_FILE_OR_DIR> | --bam <BAM_FILE_OR_DIR>] --out <DIR>
[--ref <TSV> | --gff <GFF> | --busco <FULL_TABLE.TSV> | --all]
Mandatory:
Coverage data (choose one)
--cov STR Coverage file (COV) created by construct_cov_file.py or directory containing
multiple coverage files
--bam STR BAM file to automatically create the coverage file
Output directory
--out STR Output directory
Reference regions to calculate average coverage (choose one)
--ref STR File containing TAB-separated chromosome, start, and end
--gff STR GFF3 file containing genes given by BUSCO
--busco STR BUSCO annotation file (full_table_busco_run.tsv)
--all Use all positions of the assembly
Optional:
--black STR Sequence ID list for exclusion
--gzip Search for files "*cov.gz" in --cov if this is a directory
--bam_is_sorted Do not sort BAM file
--samtools STR Full path to samtools (if not in your $PATH)
--bedtools STR Full path to bedtools (if not in your $PATH)
--name STR Prefix for output files []
--m INT Samtools sort memory [5000000000]
--threads INT Samtools sort threads [4]
--plot TRUE|FALSE Activate or deactivate generation of figures via matplotlib[FALSE]
--blackoff TRUE|FALSE Deactivate the black listing of contigs with high coverage values [FALSE]
WARNING:
- MGSE requires absolute paths (at least use of absolute paths is recommended)
- Per default contigs with very high coverage values are put on a black list to prevent inflation of the genome size prediciton by plastome contigs (in plants). However, this function can be disabled via --blackoff to estimate genome sizes with more fragmented assemblies.
Possible reference regions:
-
--refA very simple TAB-separated text file with information about chromosome, start, and end of regions which should be used as a reference set for the coverage calculation. -
--gffA GFF3 file with genes which should serve as reference regions. Only the GFF3 file produced by BUSCO can be used here. -
--buscoThis will extract the single copy BUSCOs from the provided TSV file. -
--allAll positions of the assembly will be included in the average coverage calculation.
Usage:
python construct_cov_file.py
Mandatory:
--in STR Bam file
--out STR Output file
Optional:
--bam_is_sorted Do not sort BAM file
--m INT Samtools sort memory [5000000000]
--threads INT Samtools sort threads [4]
Pucker B. Mapping-based genome size estimation. bioRxiv 607390; doi: https://doi.org/10.1101/607390