cellHarmony-Align
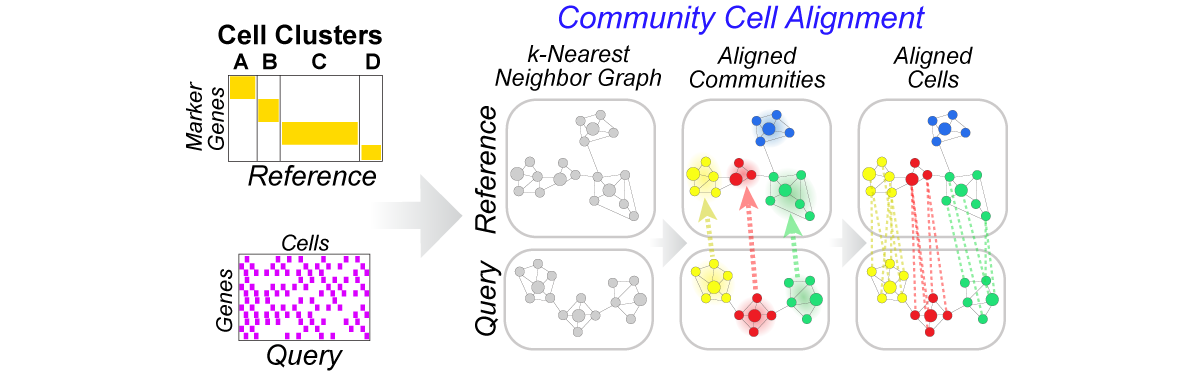
Python library for community-clustering-based label projection from a reference to query scRNA-Seq dataset (h5, mtx, txt or csv) to find cells in the reference that are most similar to cells in query. Each CellRanger h5 file is read and partitioned by clustering on the k-nearest neighbor graph. The h5 files can be restricted to marker genes (recommended) and annotated with a custom labels file (recommended). The GUI version of the program with additional functionality can be found at: http://www.altanalyze.org. More information can be found at http://www.altanalyze.org/cellHarmony/.
Note, if the data is provided as a sparse (MM) .mtx file not contained in an .h5 file, separate feature and barcode name files (genes.tsv and barcodes.tsv or .gz versions of each, formatted as per cellRanger output) must be provided in the same directory as the expression data.
Authors
Phillip Dexheimer (CCHMC), Nathan Salomonis (CCHMC), AltAnalyze team
Requirements
python 2.7 scipy numpy annoy python-louvain networkx h5py gzip
Usage
python scr/cellHarmony_align.py reference/GSM3489185_Donor_02_h5.h5 query/GSM3489183_IPF_01_h5.h5 alignments.txt --genes genes/markers.txt --labels labels/CustomLabels.txtusage: cellHarmony_align.py [-h] [-g GENES] [-s GENOME] [-k NUM_NEIGHBORS]
[-t NUM_TREES] [-l LOUVAIN] [-m MIN_CORRELATION]
[-b LABELS]
reference_h5 query_h5 output
positional arguments:
reference_h5 a CellRanger h5 file
query_h5 a CellRanger h5 file
output the result file to write
optional arguments:
-h, --help show this help message and exit
-g GENES, --genes GENES
a tab-delimited text file with genes in the first column. Also compatible with ICGS heatmap results.
-s GENOME, --genome GENOME
genome aligned to (optional)
-k NUM_NEIGHBORS, --num_neighbors NUM_NEIGHBORS
number of nearest neighbors to use in clustering,
default: 10
-t NUM_TREES, --num_trees NUM_TREES
number of trees to use in random forest for
approximating nearest neighbors, default: 100
-l LOUVAIN, --louvain LOUVAIN
what level to cut the clustering dendrogram. 0 is the
most granular, -1 the least. Default: 0
-m MIN_CORRELATION, --min_correlation MIN_CORRELATION
the lowest correlation permissible between clusters.
Any clusters in query that don't correlate to ref at
least this well will be skipped. Default: -1
-b LABELS, --labels LABELS
a tab-delimited text file with two columns (reference
cell barcode and cluster name)