

Selma is a whole genome (germline) variant calling workflow developed at the University of Bergen based on the GATK suite of tools. The guiding philosophy behind it is that it should be easy to setup, easy to use and that it utilizes system resources efficiently. This is achieved by adopting a user centric frame of mind that aims to simplify complex tasks without sacrificing functionality. The workflow itself is based on Snakemake and all dependencies are handled by using Docker and Singularity container technology. The current intended platform is TSD but support for HUNT-cloud as well as local execution is planned for future releases.
Selma is named after the mythical Norwegian sea serpent that supposedly lives in Lake Seljord
The workflow development is currently supported by Elixir2, NorSeq and Tryggve2, and in the past also by BioBank Norway.
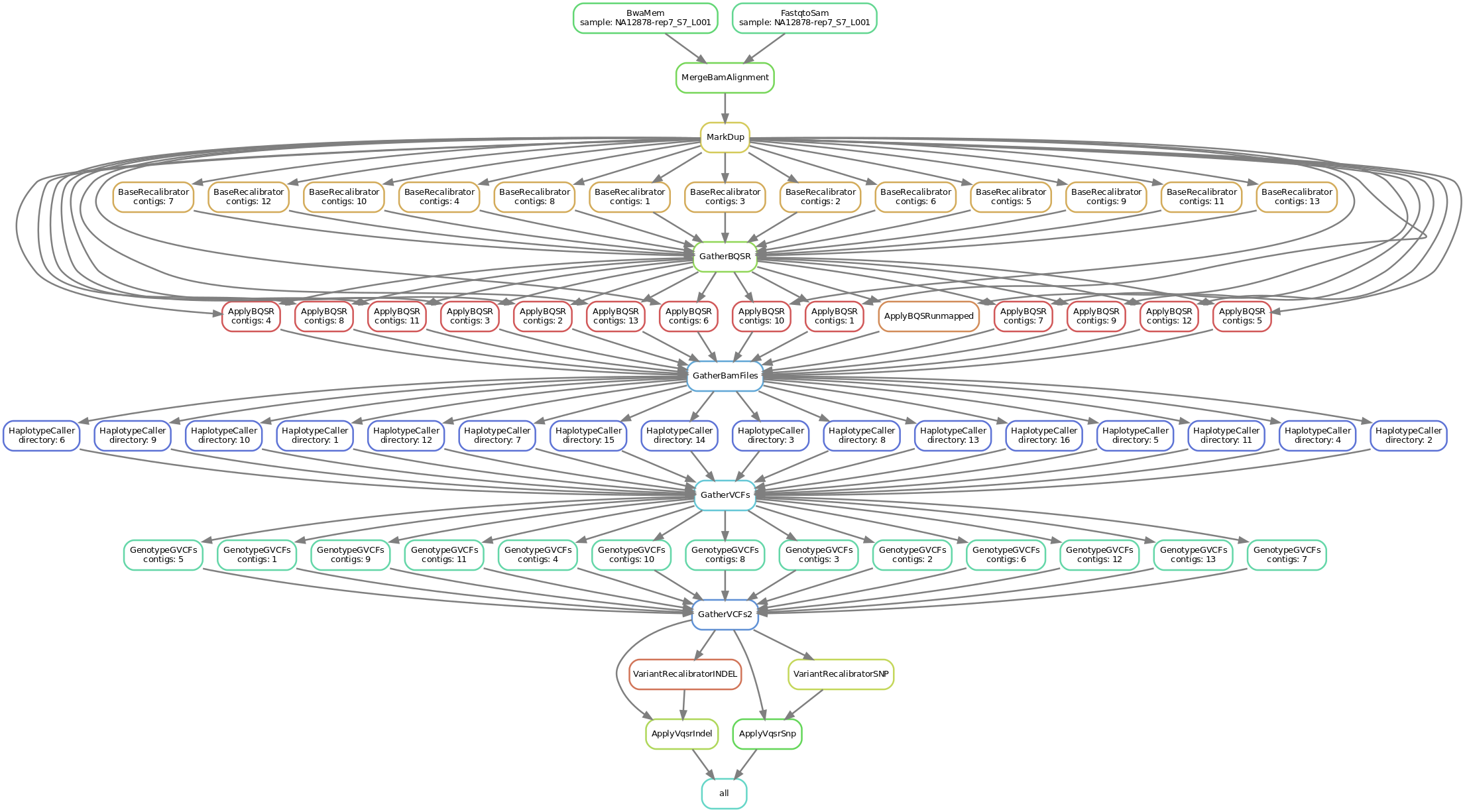
This is a simplified graph portraying the key steps that the workflow goes through, this is a complete overview including every single step. The steps that have been left out only perform "administrative" functions and don't add to the data analysis per se.
bwa version 0.7.15-2+deb9u1 - Maps fastq file to reference genome
samtools version 1.3.1-3 - bwa pipes its output to samtools to make a bam output file
The following tools are all gatk version 4.1.2.0
SplitIntervals - Splits interval list for scatter gather parallelization
FastqToSam - Converts fastq files to unmapped bam files
MergeBamAlignment - Merge aligned BAM file from bwa with the unmapped BAM file from FastqToSam
MarkDuplicates - Identifies duplicate reads
BaseRecalibrator - Generates recalibration table for Base Quality Score Recalibration
GatherBQSRReports - Gather base recalibration files from BaseRecalibrator
ApplyBQSR - Apply base recalibration from BaseRecalibrator
GatherBamFiles - Concatenate efficiently BAM files from ApplyBQSR
HaplotypeCaller - Call germline SNPs and indels via local re-assembly of haplotypes
GenotypeGVCFs - Perform genotyping on one pre-called sample from HaplotypeCaller
VariantRecalibrator - Build a recalibration model to score variant quality for filtering purposes
ApplyVQSR - Apply a score cutoff to filter variants based on a recalibration table
Supervisor
Kjell Petersen
Main developer
Oskar Vidarsson