![]()
Chemplot is a python library for chemical space visualization that allows users to plot the chemical space of their molecular datasets. Chemplot contains both structural and tailored similarity algorithms to plot similar molecules together based on the needs of users. Moreover, it is easy to use even for non-experts.
| Version | Downloads | License | Documentation | Testing |
|---|---|---|---|---|
 |
 |
 |
You can find the detailed features and examples in the following link: User Manual.
ChemPlot is also available as a web application. You can use it at the following link: Web Application.
You can find the details for the background on ChemPlot in our paper. You can download our paper at: Paper.
There are two different options to install ChemPlot:
To install ChemPlot using conda, run the following from the command line:
conda install -c conda-forge chemplot
ChemPlot requires RDKit, which cannot be installed using pip. The official RDKit installation documentation can be found here.
After having installed RDKit, ChemPlot can be installed using pip by running:
pip install chemplot
ChemPlot is a cheminformatics tool whose purpose is to visualize subsets of the chemical space in two dimensions. It uses the RDKit chemistry framework, the scikit-learn API and the umap-learn API.
To demonstrate how to use the functions the library offers we use BBBP (blood-brain barrier penetration) [1] molecular dataset. BBBP is a set of molecules encoded as SMILES, which have been assigned a binary label according to their permeability properties. This dataset can be retrieved from the library as a pandas DataFrame object.
import chemplot as cp
data_BBBP = cp.load_data("BBBP")
To visualize the molecules in 2D according to their similarity it is
first needed to construct a Plotter object. This is the class
containing all the functions ChemPlot uses to produce the desired
visualizations. A Plotter object can be constructed using
classmethods, which differentiate between the type of input that is feed
to the object. In our example we need to use the method from_smiles. We
pass three parameters: the list of SMILES from the BBBP dataset, their
target values (the binary labels) and the target type (in this case “C”,
which stands for “Classification”).
plotter = cp.Plotter.from_smiles(data_BBBP["smiles"], target=data_BBBP["target"], target_type="C")
When the Plotter object was constructed descriptors for each SMILES
were calculated, using the library
mordred,
and then selected based on the target values. We reduce the number of
dimensions for each molecule from the number of descriptors selected to only 2.
ChemPlot uses three different algorithms in order to achieve this.
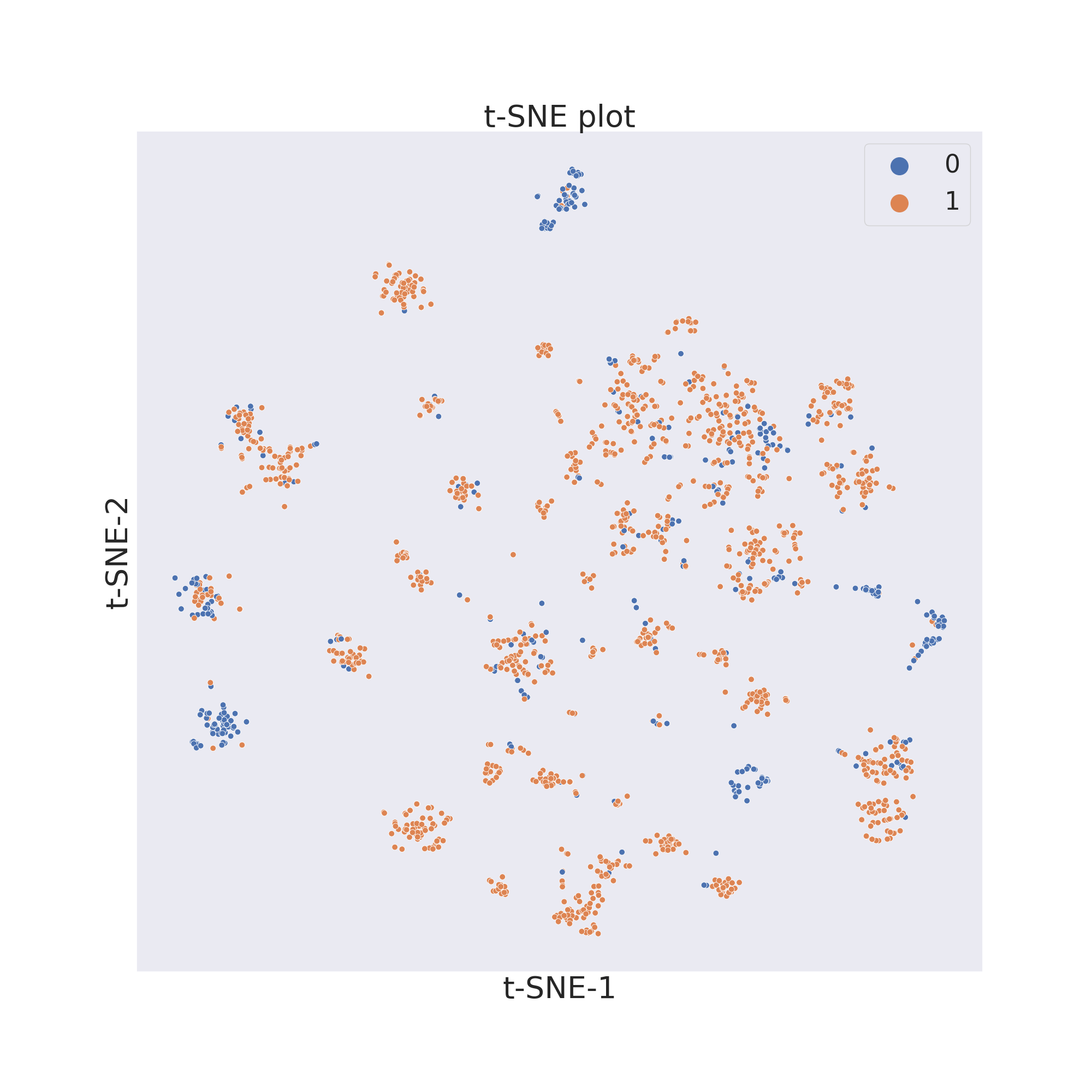
In this example we will first use t-SNE [2].
plotter.tsne()
The output will be a dataframe containg the reduced dimensions and the target values.
| t-SNE-1 | t-SNE-2 | target |
|---|---|---|
| -41.056122 | 0.355575 | 1 |
| -35.535915 | 21.648867 | 1 |
| 23.771597 | -14.438373 | 1 |
To now visualize the chemical space of the dataset we use visualize_plot().
plotter.visualize_plot()
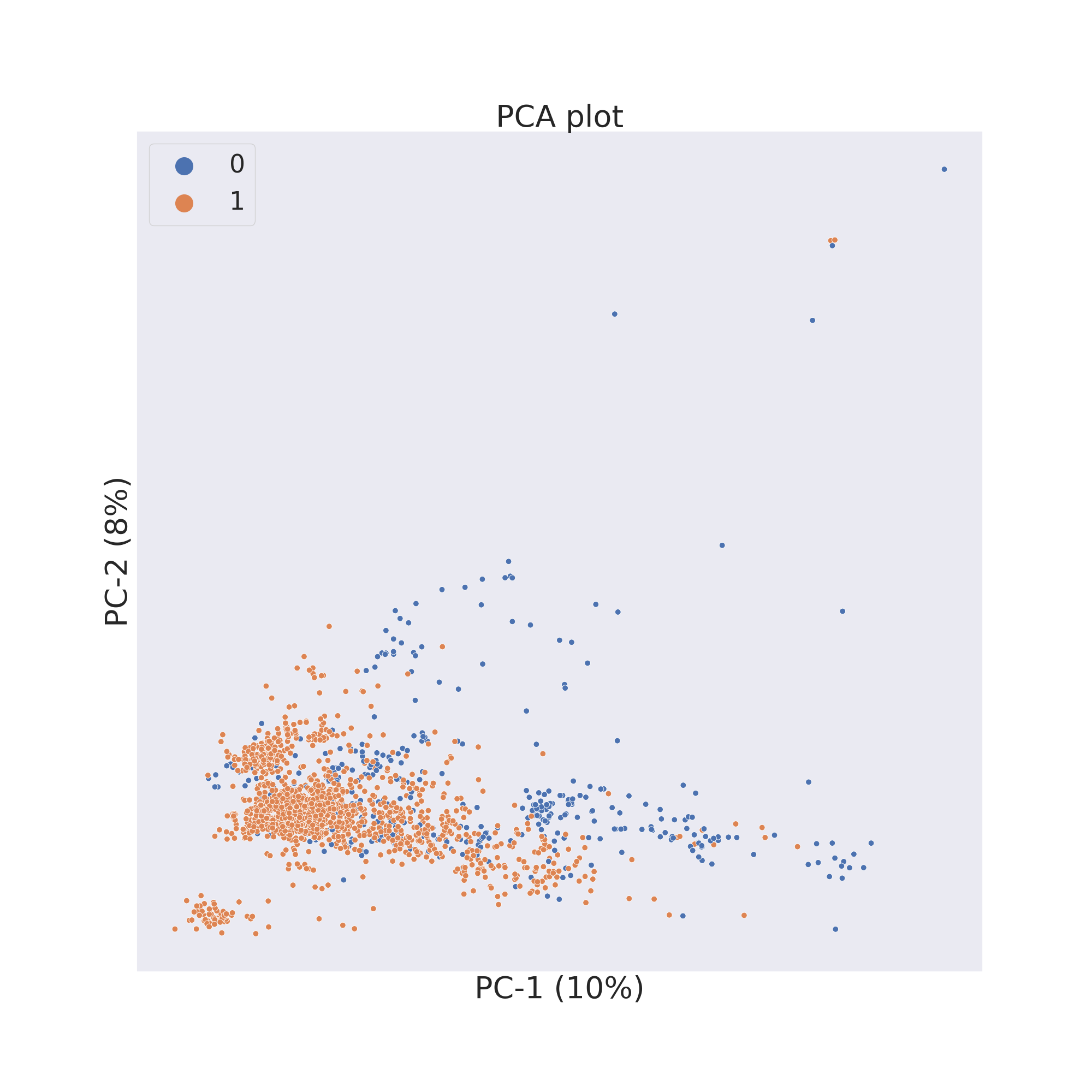
The second figure shows the results obtained by reducing the dimensions of features Principal Component Analysis (PCA) [3].
plotter.pca()
plotter.visualize_plot()
The third figure shows the results obtained by reducing the dimensions of features by UMAP [4].
plotter.umap()
plotter.visualize_plot()
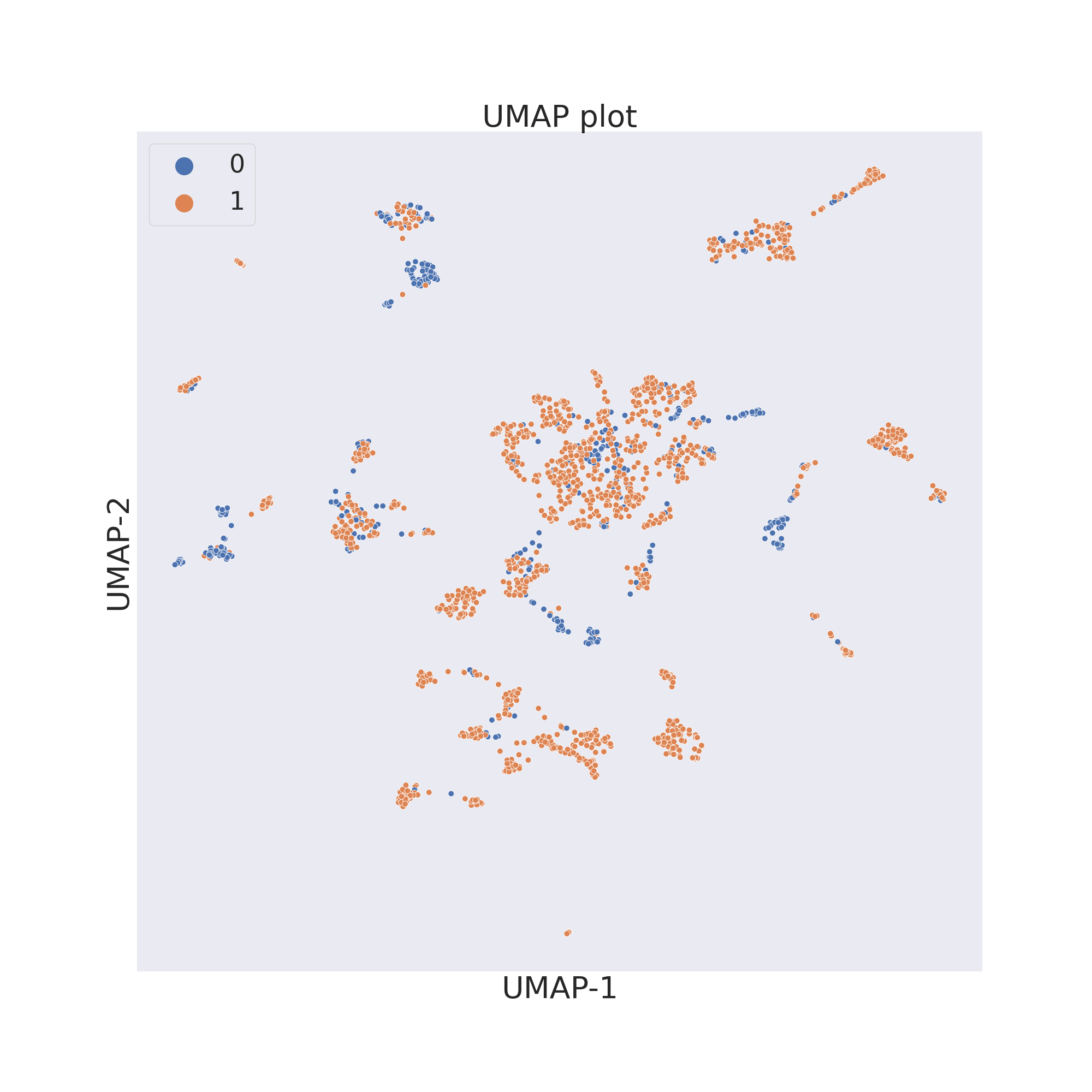
In each figure the molecules are coloured by class value.
If you use ChemPlot for your scientific projects, we would appreciate if you would cite the paper from the Chemestry-Methods journal:
@article{2022ChemPlot,
author = {Cihan Sorkun, Murat and Mullaj, Dajt and Koelman, J. M. Vianney A. and Er, Süleyman},
title = {ChemPlot, a Python Library for Chemical Space Visualization},
journal = {Chemistry–Methods},
volume = {2},
number = {7},
pages = {e202200005},
keywords = {chemical space visualization, cheminformatics, molecular similarity, Python, tailored similarity},
doi = {https://doi.org/10.1002/cmtd.202200005},
url = {https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/cmtd.202200005},
eprint = {https://chemistry-europe.onlinelibrary.wiley.com/doi/pdf/10.1002/cmtd.202200005},
abstract = {Visualizing chemical spaces streamlines the analysis of molecular datasets by reducing the information
to human perception level, hence it forms an integral piece of molecular engineering, including chemical library design,
high-throughput screening, diversity analysis, and outlier detection. We present here ChemPlot, which enables users to
visualize the chemical space of molecular datasets in both static and interactive ways. ChemPlot features structural and
tailored similarity methods, together with three different dimensionality reduction methods: PCA, t-SNE, and UMAP.
ChemPlot is the first visualization software that tackles the activity/property cliff problem by incorporating tailored similarity.
With tailored similarity, the chemical space is constructed in a supervised manner considering target properties. Additionally,
we propose a metric, the Distance Property Relationship score, to quantify the property difference of similar (i. e. close)
molecules in the visualized chemical space. ChemPlot can be installed via Conda or PyPI (pip) and a web application is freely
accessible at https://www.amdlab.nl/chemplot/.},
year = {2022}
}For any question you can contact us through email:
[1]: Martins, Ines Filipa, et al. (2012). A Bayesian approach to in silico blood-brain barrier penetration modeling. Journal of chemical information and modeling 52.6, 1686-1697
[2]: van der Maaten, Laurens, Hinton, Geoffrey. (2008). Viualizingdata using t-SNE. Journal of Machine Learning Research. 9. 2579-2605.
[3]: Wold, S., Esbensen, K., Geladi, P. (1987). Principal component analysis. Chemometrics and intelligent laboratory systems. 2(1-3). 37-52.
[4]: McInnes, L., Healy, J., Melville, J. (2018). Umap: Uniform manifold approximation and projection for dimension reduction. arXivpreprint arXiv:1802.03426.