Improving convergence for implicit solvation
Closed this issue · 5 comments
Hello dear Shankar,
I am asking for your help in setting up proper parameters for implicit solvation calculations.
I am trying to compare energies for both vacuum and implicit solvated Na [100] slabs and calculate surface energy of interaction of metal surface with fluid. I started from the simplest linearPCM GLSSA13.
I have found that solvated calculations run much slower on production, and therefore tried to do first a 3 layer test slab for training. I do ionic-minimize for both vacuum and solvated calculations in order to find accurate values of energy.
I attach output of still running calculations.
Vacuum calculation runs smoothly (the first run of attached X.out file).
Unfortunately linearPCM with EC solvent is much slower. I understand that there is no free lunch in scientific calculations, and, therefore, ElecMinimize time increase from 24 to 32 secs is ok. Actually, problem arises with convergence.
I see several issues with convergence I want to mitigate:
- |grad|_K oscillates strongly
- I often encounter messages "Wrong curvature in test step " or ""Undoing step", indicating dificult covergence
- I often encounter "IonicMinimize: Wolfe criterion not satisfied" messages, indicating not very good prediction of next model.
I see them for implicit solvation very often.
I encounter "Wolfe criterion not satisfied" with vacuum too when doing ionic-minimize or lattice-minimize with
energyDiffThreshold 1e-08 for ElecMinimize,
energyDiffThreshold 1e-07 and knormThreshold 1e-05 for ionic-minimize or lattice-minimize .
Actually knormThreshold 1e-05 significantly improves outcome when doing surface energy calculations.
Also I see significant vertical expansion between layers, say thickness increases from 4.207 to 4.27Å (see attached Vesta file).
The energy of liquid-metal interaction looks quite high and is at least -3J/m2. I don't know, whether these are reasonable values.
https://github.com/ximik69/JDFTx_debug_output/raw/main/Na_100_solvation_debug/3xNa_100_EC.out
Could you please tell me the optimal settings I should use?
Best wishes,
Igor.
Hi Igor,
I strongly suspect you are encountering 'leakage' of fluid into the electronic system, which is a particular problem of the local solvation models like GLSSA13 (which is what later became VASPsol) that you are using. Note that your A_diel energy component is over 6 Hartrees, which is what leads me to this conclusion. For a neutral metal system, this should be a few milliHartrees. This numerical issue is likely causing your slow electronic convergence.
I know you are forced to use this local solvation model because CANDLE, SaLSA etc. are not parameterized for EC. However, just as a point of comparison, run the same system with CANDLE water and let's look at the numbers for that. We can assume that the water solvation energy should be an upper bound for what we could expect for EC.
Best,
Shankar
Dear Shankar,
thank you very much for your help.
I was not forced to GLSSA13 because of lack of parametrization, I tried it just to learn. I tried it as a solvent for metal-ion batteries in a hope to find some experimental data in future.
I checked fluid leakage and it is the case:
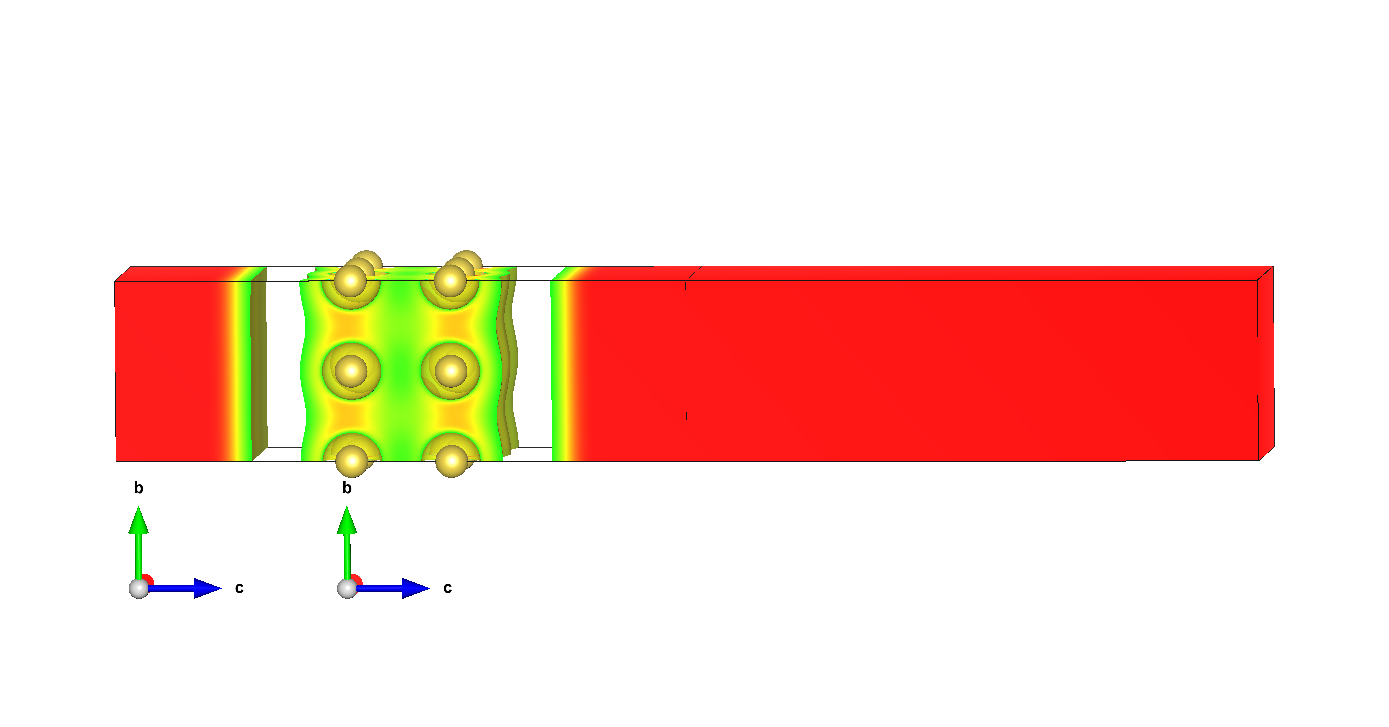
isolevel is 0.5
There are also places with isolevel 0.8 between Na atoms.
fluidShape
fluidShape
one should unpack xz archive first to open *.vesta file.
There are also bound charges around all Na atoms:
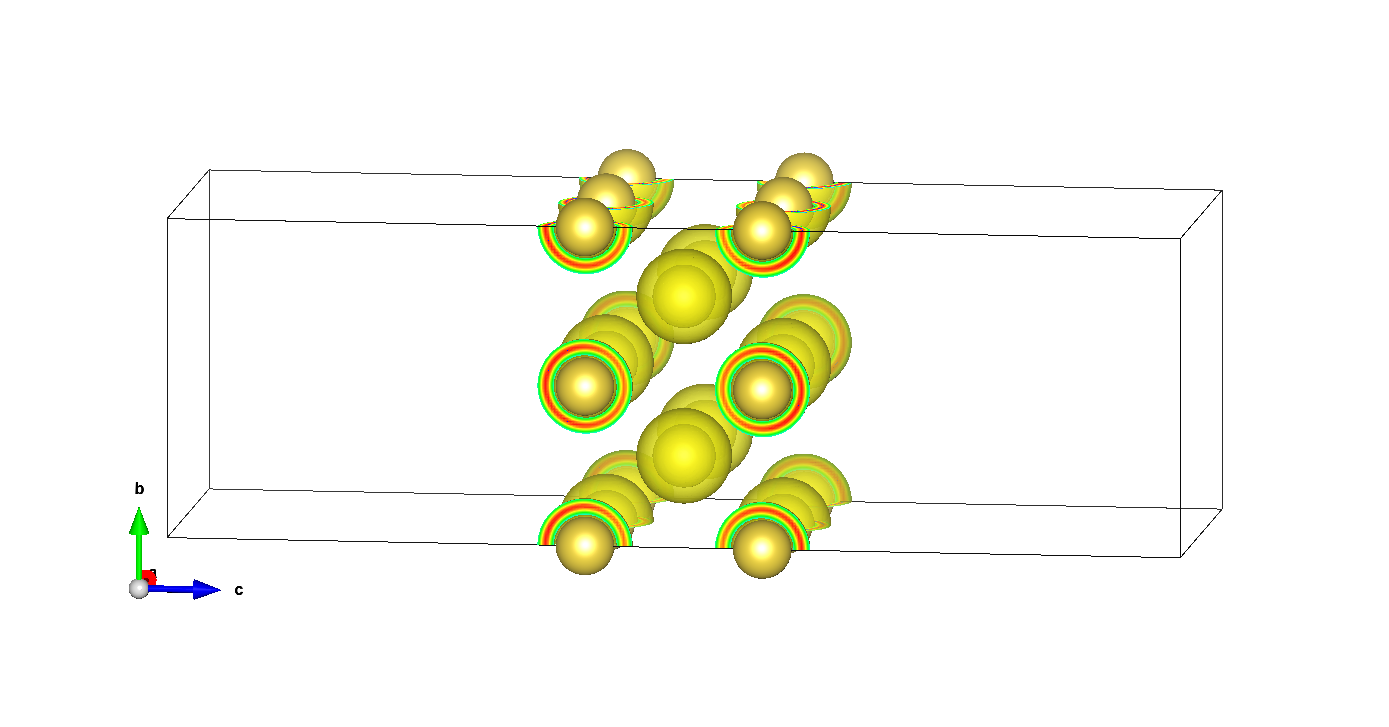
X.nbound.xsf.xz
isosurface level 0.01
I'll recalculate with water and CANDLE and compare.
Best wishes,
Igor.
Sounds good: make sure you start with a geometry where the ions are at their equilibrium spacing, so there isn't a large enough gap where even realistically water can start entering.
Hi dear Shankar,
I have tested 3-layer Na [100] slab with CANDLE fluid model for water and it seems to fix leaking problem.
I always use equilibrium structures (here 3-layer Na [100] slab in vacuum with 22.4 Å between layers without vdW interactions) for fluid in order to avoid problems. Previous fluid model showed leaking and unphysical difference between vacuum and solvated calculation.
Unfortunately another problem arises - it looks like interaction of sodium surface with water is slightly hydrophobic.
for instance surface energy of surface solvation
sigma(solv)=(Esolvated-Evacuum)/(2*S)
is slightly positive.
sigma(solv)=0.001J/m2 w/o vdW interactions
sigma(solv)=0.0027J/m2 w Grimme D2 vdW interactions
sigma(solv)=0.001 J/m2 w Grimme D3 vdW interactions
This seems kinda unphysical as usually metals are wetted by water.
Could you please give me advice, what to do next?
Best wishes,
Igor.
Implicit solvation will not capture energy effects due to charge transfer and bonding between the solute and solvent. FOr those effects, an explicit solvent layer followed by implicit solvent is typically needed. This result is reasonable for non-bonded interactions alone.
Best,
Shankar