Graph Classification & Batchwise Training
rushilanirudh opened this issue · 68 comments
Hi I have two questions:
- I would like to repurpose this for graph classification (where I have a single label per graph). I see there is an option to choose
featureless=Falsewhen defining a newGraphConvolutionlayer. However, the loss is still computed for each node, and I was wondering how I should change your code. - In the context of graph classification, how should I modify your
train.pyfor batch-wise training?
Thanks for putting this together!
Thanks for your questions. For graph-level classification you essentially have two options:
- "hacky" version: you add a global node to the graph that is connected to all other nodes and run the standard protocol. You can then interpret the final value of this global node as graph-level label.
- global pooling: as a last layer, you can implement a global pooling layer that performs some form of pooling operation (optionally with attention mechanism as in https://arxiv.org/abs/1511.05493) over all graph nodes
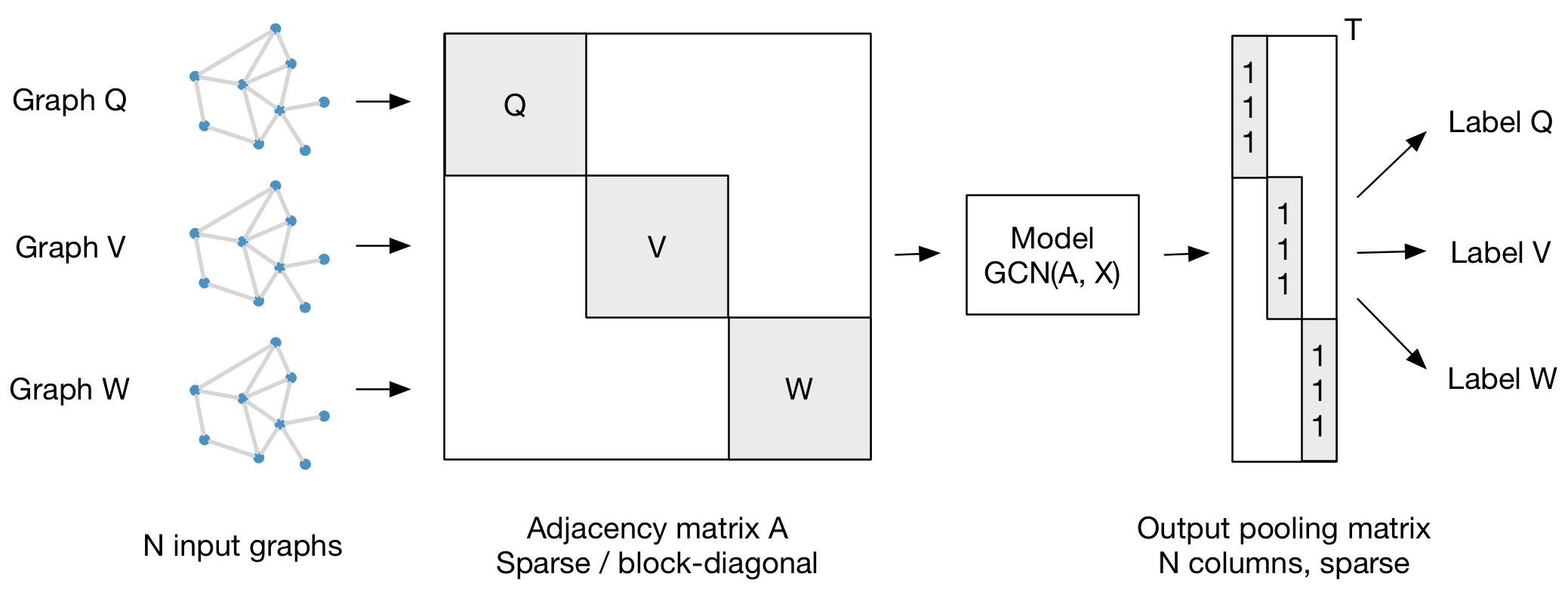
For batch-wise training over multiple graph instances (of potentially different size) with an adjacency matrix each, you can feed them in the form of a block-diagonal adjacency matrix (each block corresponds to one graph instance) to the model, as illustrated in the figure below:
Hi,
I have a followup question here. Though the code has a featureless option, I do not think it works directly when featureless is set to True. I just wanted to confirm that.
Thanks for your great code !
Hi, thanks for the catch. For the featureless mode, you have to set the input dimension of the first layer (only the first layer supports featureless) to be the number of nodes in the graph. I'll make sure to add a sentence or two about this in the documentation at some point.
Hi,
I want to use this gcn framework to do a semi-supervised regression mission (Given a graph(V, E) with feature matrix, the value(continuous) of some node is known while others are not, Target: predict the value of those unknown nodes). I directly change the loss function into RMSE, but it doesn't work well on validation dataset and test dataset.
so,
-
is GCN suitable for regression?
-
how to change this framework to meet the requirements of regression?
Thanks you !
As far as I know regression should work. But you may want to look at the A-normalization. If I remember correctly, the normalization method proposed by Kipf & Welling does not lead to the rows of the normalized adjacency summing to 1.
It might be a good idea to turn the regression problem into a classification problem by bucketing the real-valued targets into several classes. This typically (empirically) works quite well in combination with a (softmax) cross-entropy loss. See, e.g., the WaveNet paper (https://arxiv.org/abs/1609.03499) for details.
It might also work to cast the regression problem as a classification problem by augmenting the input data similar to support vector regression. For example, regression from x to y, can be converted to a classification task with data ([x, y+r], 1) and ([x, y-r], -1). Thus no need to change the normalization term.
That sounds interesting. What would 'r' be in this case?
'r' is a manually set real-value margin, e.g., r = 0.8. The setting of 'r' typically depends on the range of y (to ensure most/all features are separable) and how much you'd like the samples to be separable. See here for details, where r is the epsilon in this case.
Hi, it is not clear to me how to give to the model a batch of graphs instead of only one. Could you please give other details? Thanks
Hi @tommy9114,
If I remember correctly (last time I touched it was in April) it is not possible to forward-pass multiple graphs at once, because you'd need a rank>2 sparse Tensor. Attempts were made to support rank>2 sparse Tensors with tensorflow (tensorflow/tensorflow#9373) but it did not work out.
So if you want to pass multiple graphs at once, you should find out if any of the other frameworks (PyTorch?) support rank>2 sparse Tensors.
yea in fact I previously read about this block-diagonal sparse matrix but actually I don't really know what is it and how to create it. I didn't find anything on google about how to build such data structure.
The following figure should hopefully clarify the idea:
@tkipf Sorry to beat this into the ground. But, I'm interested in classifying about 2000 (i.e., N = 2000) 100x100 adjacency matrices (representing graphs) into two different classes -- i.e., whole-graph labelling. Going by your figure, this means that I ought to feed it a sparse 200,000 x 200,000 adjacency matrix into the model (i.e., each of the 2000 graphs represented along the diagonal). And the output pooling matrix is 200,000 x 2000, with the class labels along the diagonals (i.e., where the 1's are in your figure). Is this correct or am I completely missing something?
This is correct, but I would recommend training the model using smaller mini-batches of, say, 32 or 64 graphs each. Make sure the pooling matrix is also sparse and that you're using sparse-dense matrix multiplications wherever possible. This should run quite fast in practice. Good luck!
@tommy9114 Same nodes and number of nodes, different edge values.
Hi, does anyone successfully use this for graph classification?
I implement a toy example on mnist using each pixel as node and its intensity as feature.
However, the loss did not go down?
I have modified the graphconv layer to dense matrix to work with parallel data loader.
And the "ind" (size: N*batch, values are normalized to the number of nodes in each graph) is the pooling matrix as in the figure
class GCN(nn.Module):
def __init__(self, dropout):
super(GCN, self).__init__()
self.gc1 = GraphConv(1, 16)
self.gc2 = GraphConv(16, 32)
self.fc1 = nn.Linear(32, 500)
self.fc2 = nn.Linear(500, 10)
self.dropout = dropout
def forward(self, x, adj, ind):
# Conv layer
x = F.relu(self.gc1(x, adj))
x = F.relu(self.gc2(x, adj))
# Batching before fc layer
x = torch.mm(ind, x)
# FC layers
x = F.relu(self.fc1(x))
x = F.dropout(x, self.dropout, training=self.training)
x = self.fc2(x)
return x
I used this model quite some time ago for graph-level classification of the datasets provided in this paper: https://arxiv.org/abs/1605.05273 and got comparable (sometimes worse, sometimes better) results compared to their method. So maybe just have a look at these datasets. Shouldn't be too hard to load them in and run the model on these.
In order to go from node-level representation to a graph-level representation you will indeed have to perform some kind of order-invariant pooling operation. Any popular pooling strategy typically works fine, in my experience: attentive pooling (https://arxiv.org/abs/1703.03130) often works better than max pooling which in turn often works better than average pooling. Make sure to allocate a somewhat large representation size for the pre-pooling layer (as you're going to average a lot of values). You can also pool the representation of every layer individually and concatenate or add/average the result (similar to https://arxiv.org/abs/1509.09292).
If you don't have any initial node features, simply use an identity matrix (assuming the identity and position of every node is the same across all the graph samples) as an initial feature matrix. This essentially provides a unique one-hot feature vector for every node in the graph. Only works if node identities stay the same across all graph samples. Otherwise you will have to come up with some sensible feature representation (e.g. node degree, betweenness centrality, etc.).
Sorry to keep asking question but I'm not sure if I got the right intuition:
The architecture is able to learn feature vectors for nodes only if I pass always the same graph right? If my training set has different graphs (assume they have all the same dimension) but each row in the adj matrix may represents a different node then this architecture is not a good fit to learn the fetures of the nodes right?
Thanks for your patient and kind answers
No worries! Your intuition is correct in the absence of any node features, i.e. if you pick an identity matrix for the initial feature matrix. If, however, you describe nodes by features (from a feature space which is shared by all nodes, i.e. two nodes could have the same features) the model can learn from different graphs (e.g. molecules), such as done here: https://arxiv.org/abs/1509.09292
Yes, sure! https://arxiv.org/abs/1509.09292 uses node degree as an initial feature, for example.
Hello,
@rushilanirudh @tkipf have you succeeded to update the code to deal with graph classification (rather than node classification. Each graph has only one class)?
Thank you
I do not intend to release this implementation as part of this repository. But it shouldn't be too difficult to implement this yourself :-)
Edit: PRs are welcome
I've implemented a self-attention layer:
class SelfAttention(Layer):
def __init__(self, attention_dim, bias_dim, hidden_units, **kwargs):
super().__init__(**kwargs)
self.hidden_units = hidden_units
self.A = None
self.vars['Ws'] = tf.Variable(tf.random_uniform([attention_dim, self.hidden_units]))
self.vars['W2'] = tf.Variable(tf.random_uniform([bias_dim, attention_dim]))
def _call(self, inputs):
aux = tf.tanh(tf.matmul(self.vars['Ws'], inputs, transpose_b=True))
self.A = tf.nn.softmax(tf.matmul(self.vars['W2'], aux))
tf.summary.histogram('self_attention', self.A)
out = tf.matmul(self.A, inputs)
out = tf.reshape(out, [out.get_shape().as_list()[0] * out.get_shape().as_list()[1]])
return out
you can stack it on top of the gc layers. (Not sure if it is 100% correct, if you find something wrong please tell me!)
Best,
@tommy9114 Thank you for you answer. It looks ok (but l confirm when testing).
So have you modified the loss function so that the loss is computer on the whole graph rather than at each node ?
The attention layer take all the nodes embeddings into account and build a weighted average (where the weights are learnt) of those outputing a single hidden vector that should represent the whole graph. You can stack another dense layer with softmax to do the classification
So as a last layer on the top of graph convolutional layers you add an attention layer (equation 7 in https://arxiv.org/pdf/1511.05493.pdf) then we add a dense layer with softmax to do classification. This process allows to keep the loss function (primarily defined to be applied at node level ) for graph classification.
Correct me if l'm wrong !
I don't know the paper you cited (thanks for the pointer it looks very interesting). I don't think we can keep the same loss function because it was developed to deal with multiple classifications (each node had his hown prediction). Now we have only one prediction so we can use a classical cross-entropy function (i think)
l thought we talk about the same attention layer mechanism. So for you, what did you mean by the attention layer (without the pointer l gave), is it implemented here ?
I used self-attention implemented here https://arxiv.org/pdf/1703.03130.pdf as @tkipf suggested in som post above i think
@tommy9114 okey. So, to recap , adding attention layer on the top of GC (as you just mentionned) followed by a dense layer with softmax allows to keep unchanged the loss implemented to do graph classification ?
Definitely, softmax cross entropy will do the job
@tommy9114 , l'm wondering why there is no pooling layer in this graph convolutional network ?
@pinkfloyd06 Pooling on graphs hasn't been convincingly shown yet to help in predictive performance. With pooling I mean hierarchical pooling, i.e. forming a hierarchy of coarser graphs. That's why we don't consider it here for simplicity. Using global pooling (like the implementation by @tommy9114 ) is still a very good idea, though.
@tkipf Thank you for your answer. l have a follow up questions related to your comment.
- How do you explain the fact of not using any pooling layer and your network performs ?
- How do you explain that a hierarchy of coarser graph pooling doesn't help in predictive performance ? Is it related to the hierarchy itself or the way we build this hierarchy (adding fake node) ?
- l would be very happy to give me a pointer to global pooling and its implementation on a graph . Thank you for the idea , l didn't heard about it before (global pooling on graph) @tommy9114
This is getting off-topic. For global pooling the pointer by @tommy9114 is a good start: https://arxiv.org/pdf/1703.03130.pdf . Hierarchical pooling typically doesn't work well because you have to define some heuristic structure-based fixed pooling operation ahead of time. What kind of coarsening strategy works well will be highly domain dependent. There's currently no silver bullet that works well for all kinds of graph data.
@tkipf thank you for your answer. It seems to be a good idea to apply a global pooling.
l would like to hear from @tommy9114 about that and his global pooling implementation on graphs.
Hi, I have two questions:
- When I want to use batch-wise training, is it right that I can't give to the model a batch of graphs(and y), unless I concatenate the all the X together and make a sparse block matrix A as input (just treat them as a big graph)?
- When I use your method(a sparse block A), I think I should concatenate my X and y in format of (M+...+Z) *FeatNum and (M+...+Z)*classNum, and A is a sparse block matrix. Could you tell me if I am right and give some guidance?
Thanks
Yes, this is correct.
If you do not need sparse matrix operations (in case your graphs are quite small), you can resort to a much simpler implementation by creating an adjacency tensor of shape BxNxN where B is the batch size and N is the maximum number of nodes a graph has in this batch. You will need to add row- and column-wise zero-padding if you have graphs of different size in a single batch. The form of the updates for the GCN model in this case should be easy to derive. Hope this helps!
Could you please explain in more details about implementation of graph classification? Thank you!
Hi @tkipf , first of all I find this work to be really interesting. My apologies in advance for asking yet another question on how to adapt your code for graph classification. I also understand that my you may not be that familiar with the code anymore and that my question might be too precise for you to provide help.
I am relatively new to machine learning and I'm trying to implement your code to classify molecules (graphs of different sizes) into 2 classes (toxic or non-toxic). I have been working on this project for a few weeks now taking suggestions from the comments above but regardless of what I tried the model terminates early after 12 epochs with an accuracy no higher than 60%. I believe the way I am pre-processing or formatting my data is where I am going wrong as I'm trying to use the model 'off the shelf'.
I have 100 graphs (different number of nodes per graph) with 50 graphs for my 2 classes (Once my model is working I plan on using the full dataset of 1000 graphs). I'm trying to implement the 'hacky' version you mentioned at the beginning of this post by introducing a super node which is connected to all other nodes in a graph and initialized with a feature vector of 0s. I formatted my data such that each node in a graph has the same label as the graph (so if a graph is meant to be part of class 1, all nodes in that graph have the label of class 1).
I'm now unsure how I should separate my data as training features with labels, training features without labels and the test set (obtaining x, tx and allx in the code). I split my data as 60% training set and 40% testing set, with an even spread over both classes. In the training set half of the super nodes are not given a label so that I can still train the model in a semi-supervised fashion. Unless I am going about this the wrong way and I should edit the code to work for supervised-learning.
Even though I am not training the model batch-wise I assume I still have to use the block-diagonal adjacency matrix as described in the image you posted before? I also modified the way the accuracy is calculated by comparing the label prediction of the unlabeled super nodes with the label they should be predicted as.
Again sorry for the long post ... and thanks in advance for any help!
Hi @tkipf, my task is that I have a dataset {(Ai, Xi, yi)}, where Ai is adj matrix, Xi input features over some nodes, yi is the label of (Ai, Xi). That is to say Ai and Aj may be different both in nodes ,edges and dimensions(take web graph for example: Xi may represent a group of users in U.S., while Xj stands for a group of users in U.K.. yi is the label for anomaly detection of a single graph. All datas are from the same web.). I wonder if it works using GCN you propose. In your case, there's only one graph for the whole dataset. I read comments above and think this might work. However, as to my understanding, parameters in a GCN should fit the structure information of a unique graph. So how could it work if the graph varies with each datapoint? Thanks for explaining.
Hi @TZWwww ,
Yes you can have different graph structure (different adjacency matrix , variable number of nodes) for each example. But the dimensionality of your features nodes should be the same.
For practical consideration :
- Make zero padding to get adjacency matrices into the same dimension that apply a mask on each graph.
- For features dimension , it's preferable to get them into the same dimension (even if you can make a mask and zero padding as in 1) ) . But you can apply PCA and control the number of dimensions to keep.
Hope it helps
Thanks very much @ahmedmazariML for your response 👍, and it helps a lot :
Sure the feature dimension should be the same. I still have two questions:
1.Can I implement batch-wise training with adj matrix into tensor with dimension [B, m, m] where B is the batch size, m is the maximum number of nodes in that batch(some column could be padding)?
2.Why would this work? Since parameters in GCN should contain the information of a unique graph, do varied graphs leads to non-convergence?
Thanks for your patience. hh
-
Yes
-
Which parameter ? what do you mean by non-convergence ?
Thanks for explaining again 👍
The parameters I mean is the learnable parameters in the each layer of GCN. The non-convergence I mean is to say that GCN is mean to solve the problem of unique global adj matrix, it might lead to non-convergence if the adj matrix varies.
How can we plot ROC curves for GCN that are capable of classifying graphs? Thanks in advance.
Can we use centrality as node attributes or will the GCN automatically learn it?
Thank you so much.
Sounds like a reasonable set-up. A dataset of 50 labeled graphs is extremely small, so even if you did everything correctly I wouldn’t expect much. It’s probably better to work with datasets on the order of several thousands of samples or more. Your final adjacency matrix should be block-diagonal if the nodes are ordered according to which graph they belong to. If you don’t order them in any particular way, the only important property is that there are no connections between samples, I.e. every connected component in the batch adjacency matrix corresponds to one graph. In terms of labeling: please don’t try to follow the data structure of these .tx files etc (this is from a different paper and not a particularly good way to structure the data)- just write your own data loader and put the data in any reasonable format (e.g. csv files). You only need to put a class label on this one global node per graph and otherwise keep the others unlabeled (Just vectors of 0s will do). Make sure to only put a loss on the labeled global nodes. Hope this helps
…
On Mon 27. Aug 2018 at 23:11 MortZx @.***> wrote: Hi @tkipf https://github.com/tkipf , first of all I find this work to be really interesting. My apologies in advance for asking yet another question on how to adapt your code for graph classification. I also understand that my you may not be that familiar with the code anymore and that my question might be too precise for you to provide help. I am relatively new to machine learning and I'm trying to implement your code to classify molecules (graphs of different sizes) into 2 classes (toxic or non-toxic). I have been working on this project for a few weeks now taking suggestions from the comments above but regardless of what I tried the model terminates early after 12 epochs with an accuracy no higher than 60%. I believe the way I am pre-processing or formatting my data is where I am going wrong as I'm trying to use the model 'off the shelf'. I have 100 graphs (different number of nodes per graph) with 50 graphs for my 2 classes (Once my model is working I plan on using the full dataset of 1000 graphs). I'm trying to implement the 'hacky' version you mentioned at the beginning of this post by introducing a super node which is connected to all other nodes in a graph and initialized with a feature vector of 0s. I formatted my data such that each node in a graph has the same label as the graph (so if a graph is meant to be part of class 1, all nodes in that graph have the label of class 1). I'm now unsure how I should separate my data as training features with labels, training features without labels and the test set (obtaining x, tx and allx in the code). I split my data as 60% training set and 40% testing set, with an even spread over both classes. In the training set half of the super nodes are not given a label so that I can still train the model in a semi-supervised fashion. Unless I am going about this the wrong way and I should edit the code to work for supervised-learning. Even though I am not training the model batch-wise I assume I still have to use the block-diagonal adjacency matrix as described in the image you posted before? I also modified the way the accuracy is calculated by comparing the label prediction of the unlabeled super nodes with the label they should be predicted as. Again sorry for the long post ... and thanks in advance for any help! — You are receiving this because you were mentioned. Reply to this email directly, view it on GitHub <#4 (comment)>, or mute the thread https://github.com/notifications/unsubscribe-auth/AHAcYFJUCtmrENHkUcG3u_-4WkX0lZETks5uVG6agaJpZM4LofoM .
Sounds like a reasonable set-up. A dataset of 50 labeled graphs is extremely small, so even if you did everything correctly I wouldn’t expect much. It’s probably better to work with datasets on the order of several thousands of samples or more. Your final adjacency matrix should be block-diagonal if the nodes are ordered according to which graph they belong to. If you don’t order them in any particular way, the only important property is that there are no connections between samples, I.e. every connected component in the batch adjacency matrix corresponds to one graph. In terms of labeling: please don’t try to follow the data structure of these .tx files etc (this is from a different paper and not a particularly good way to structure the data)- just write your own data loader and put the data in any reasonable format (e.g. csv files). You only need to put a class label on this one global node per graph and otherwise keep the others unlabeled (Just vectors of 0s will do). Make sure to only put a loss on the labeled global nodes. Hope this helps
…
On Mon 27. Aug 2018 at 23:11 MortZx @.***> wrote: Hi @tkipf https://github.com/tkipf , first of all I find this work to be really interesting. My apologies in advance for asking yet another question on how to adapt your code for graph classification. I also understand that my you may not be that familiar with the code anymore and that my question might be too precise for you to provide help. I am relatively new to machine learning and I'm trying to implement your code to classify molecules (graphs of different sizes) into 2 classes (toxic or non-toxic). I have been working on this project for a few weeks now taking suggestions from the comments above but regardless of what I tried the model terminates early after 12 epochs with an accuracy no higher than 60%. I believe the way I am pre-processing or formatting my data is where I am going wrong as I'm trying to use the model 'off the shelf'. I have 100 graphs (different number of nodes per graph) with 50 graphs for my 2 classes (Once my model is working I plan on using the full dataset of 1000 graphs). I'm trying to implement the 'hacky' version you mentioned at the beginning of this post by introducing a super node which is connected to all other nodes in a graph and initialized with a feature vector of 0s. I formatted my data such that each node in a graph has the same label as the graph (so if a graph is meant to be part of class 1, all nodes in that graph have the label of class 1). I'm now unsure how I should separate my data as training features with labels, training features without labels and the test set (obtaining x, tx and allx in the code). I split my data as 60% training set and 40% testing set, with an even spread over both classes. In the training set half of the super nodes are not given a label so that I can still train the model in a semi-supervised fashion. Unless I am going about this the wrong way and I should edit the code to work for supervised-learning. Even though I am not training the model batch-wise I assume I still have to use the block-diagonal adjacency matrix as described in the image you posted before? I also modified the way the accuracy is calculated by comparing the label prediction of the unlabeled super nodes with the label they should be predicted as. Again sorry for the long post ... and thanks in advance for any help! — You are receiving this because you were mentioned. Reply to this email directly, view it on GitHub <#4 (comment)>, or mute the thread https://github.com/notifications/unsubscribe-auth/AHAcYFJUCtmrENHkUcG3u_-4WkX0lZETks5uVG6agaJpZM4LofoM .
Dear kipf,
Can I expect an accuracy of 90 percent and above using this. I took almost 2500 samples and got 82 percent accuracy. Now I am planning to increase my dataset to 5000 samples to improve my accuracy value. Will it be helpful?
Hi @tkipf ,
I took about 200 different size graphs for graph-level classification and got a good result. Now I'm wondering how to use the trained model to predict the graphs without labels. Can I use the model to predict a single graph?
Hi @tkipf , now I have a dataset consists of 10k small networks. The nodes of each network is 19 and all of them share a same adj matrix. That is to say, only the features of nodes are different among those networks. My task is to classify them into two categories. So I want to know whether the two methods ("global nodes" and "global pooling") are suitable in this case or not?
Thanks for your reply!
This might be a dumb question but, how do you incorporate the loss function for the output pooling matrix? Base off the image are the outputs one hot encodings or actual class labels?
Hi,
Your gcn batch operations are so great! It's used in the Graph Classification, but now, I want to use it in Node Classification. Do you think this is a feasible idea? Because I have a series of graphs, they have the same characteristic attribute.

Thanks for your question! You can certainly use the same form of graph batching for node-level prediction tasks as well. You just need to skip the "output pooling matrix" step and directly apply your node classifier on top of the output of GCN(A, X). The block-diagonal structure of the adjacency matrix ensures that no information flows between different graphs.