DENDRO, stands for Dna based EvolutioNary tree preDiction by scRna-seq technOlogy, is an R package, which takes scRNA-seq data for a tumor (or related somatic tissues) and accurately reconstructs its phylogeny, assigning each single cell from the single cell RNA sequencing (scRNA-seq) data to a subclone (Figure 1). Currently there is no phylogenetic reconstruction framework specifically designs for scRNA-seq dataset due to biological dropout (i.e. burstness), sequencing error, and technical dropout. DENDRO perfectly tackles this problem with a Bayesian framework (Beta-Binomial), and achieves high clustering accuracy .
In addition, before conducting a single cell RNA-seq experiment on a tumor sample, it is important to project how subclone detection power depends on the number of cells sequenced and the coverage per cell. To facilitate experiment design, we developed a tool, DENDROplan (Figure 2), that predicts the expected clustering accuracy by DENDRO given sequencing parameters. As a result, researchers can design experiment parameters, such as sequencing depth and number of cells, based on DENDROplan's prediction.
DENDRO: genetic heterogeneity profiling and subclone detection by single-cell RNA sequencing
If you have any questions or problems when using DENDRO or DENDROplan, please feel free to open a new issue here. You can also email the maintainers of the corresponding packages -- the contact information is shown under Developers & Maintainers.
Install to R/RStudio Install all packages in the latest version of R.
devtools::install_github("zhouzilu/DENDRO")If you observe error with Biobase try the following and then try reinstall.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("Biobase", version = "3.8")This DENDRO package includes two analysis tools: (1) DENDRO, a phylogenetic tree construction with real dataset such as tumor and hematopoesis scRNA-seq, and (2) DENDROplan, which help design experiment by predicting the accuracy of DENDRO cluster given inferred clonal tree structure, cell number and sequencing depth. Overall pipelines are shown below.
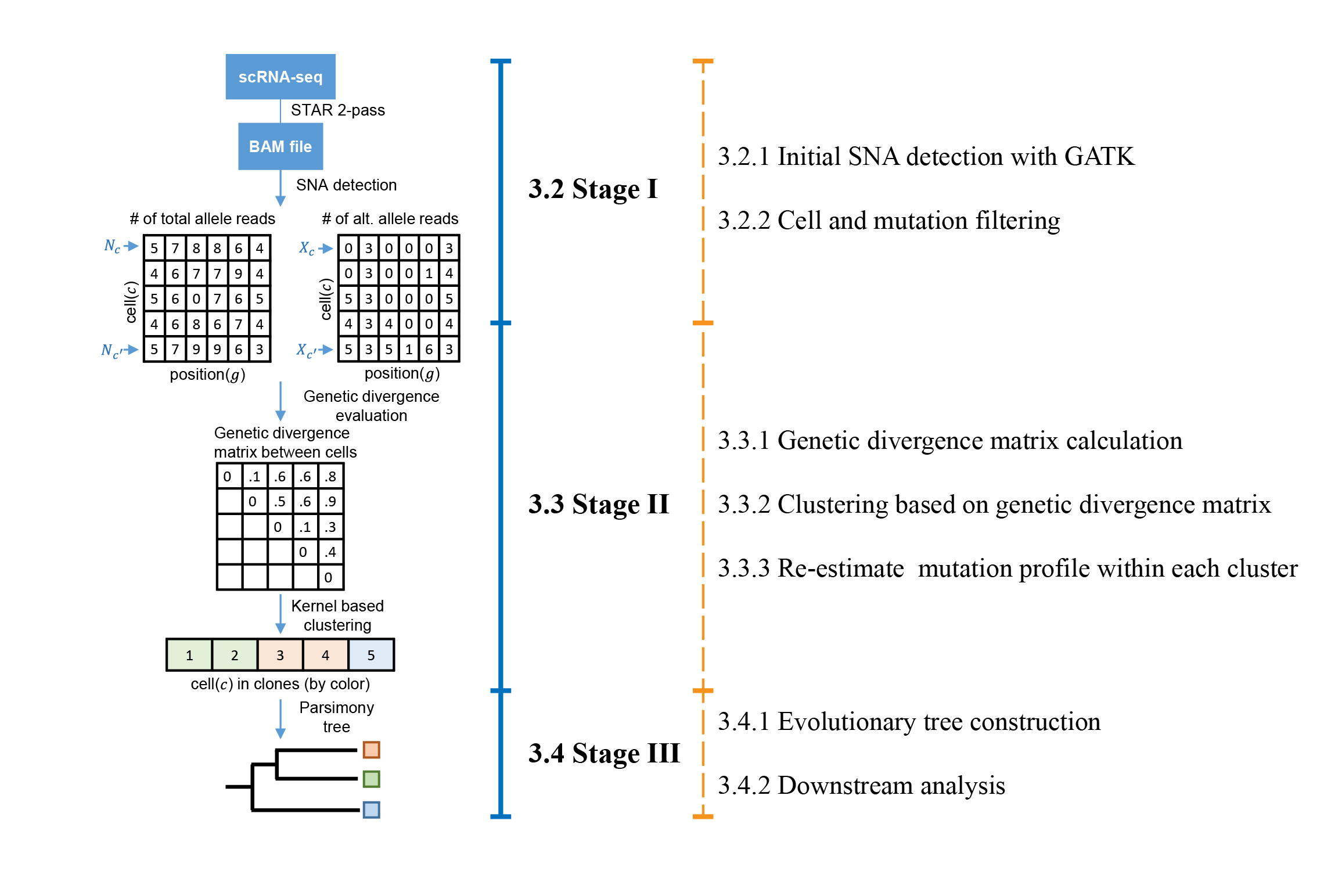
Figure 1. A flowchart outlining the procedures of DENDRO. DENDRO starts from scRNA-seq raw data. We recommend STAR 2-pass method for mapping because it is more robust with splicing junction. SNA detection was applied to mapped BAM files. Both counts of total allele reads and counts of alternative allele reads for each cell
DENDRO R notebook with step-by-step demonstration and rich display is available here. Corresponding Rmd script is available here.
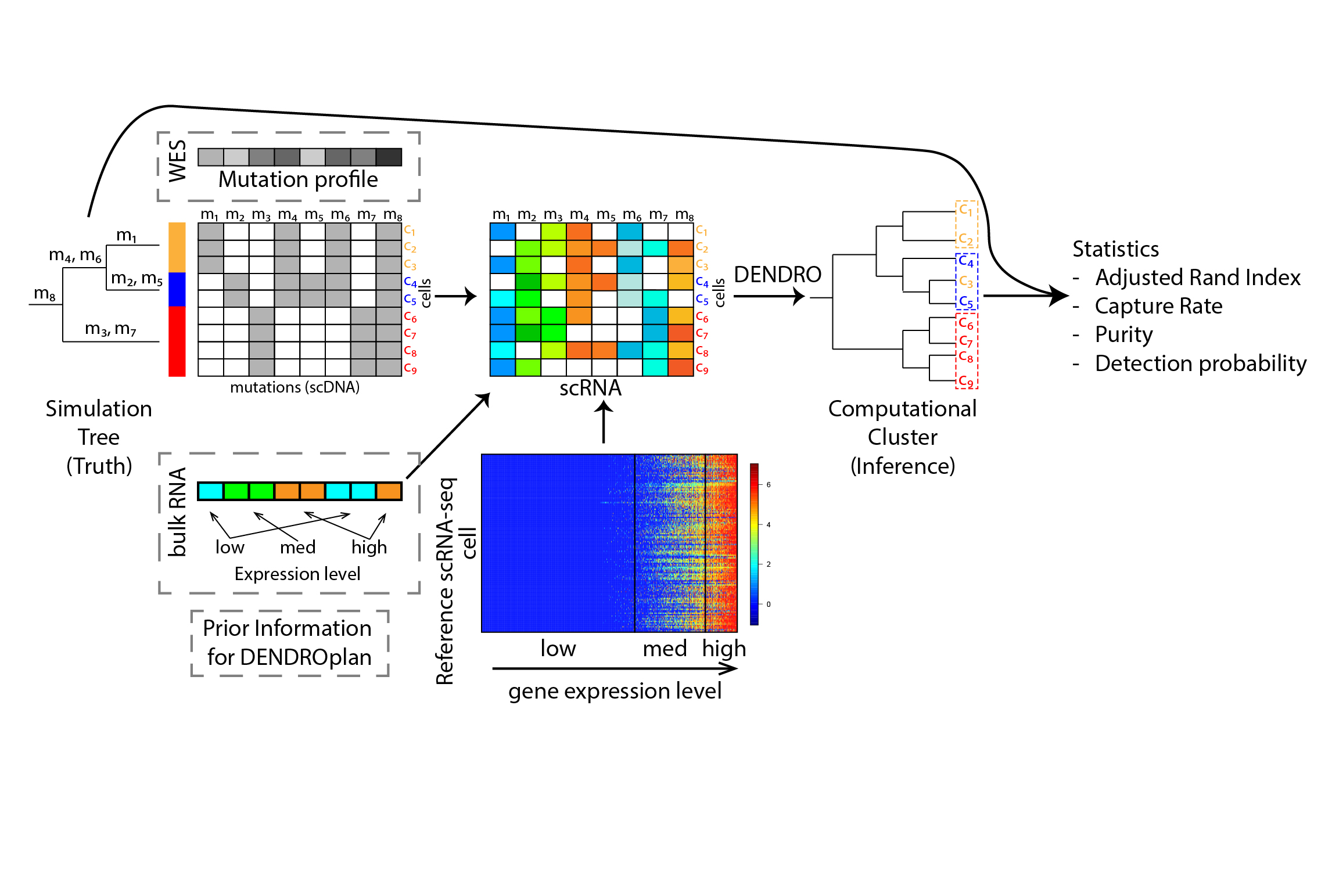
Figure 2. The overall DENDROplan pipeline. The analysis starts with a designed tree with an interested clade (purple clade in the example). Based on the tree model, number of cells, sequencing depth and sequencing error rate, we simulate single cell mutation profile. scRNA data was sampled from a reference scRNA-seq dataset given expression level in bulk. A phylogeny computed by DENDRO is further compared with underlining truth, which measured by three statistics - adjust Rand index (global accuracy statistics), capture rate (subclone specific statistic) and purity (subclone specific statistic).
DENDROplan R notebook with step-by-step demonstration and rich display is available here. Corresponding Rmd script is available here.
Zhou, Z., Xu, B., Minn, A. et al. DENDRO: genetic heterogeneity profiling and subclone detection by single-cell RNA sequencing. Genome Biol 21, 10 (2020). https://doi.org/10.1186/s13059-019-1922-x
Genetic Heterogeneity Profiling by Single Cell RNA Sequencing ([GitHub](https://github.com/zhouzilu/DENDRO))
-
Zilu Zhou (zhouzilu at pennmedicine dot upenn dot edu)
Genomics and Computational Biology Graduate Group, University of Pennsylvania -
Nancy R. Zhang
Department of Statistics, University of Pennsylvania