ABRA2 is an updated implementation of ABRA featuring:
- RNA support
- Improved scalability (Human whole genomes now supported)
- Improved accuracy
- Improved stability and usability (BWA is no longer required to run ABRA although we do recommend BWA as the initial aligner for DNA)
ABRA2 requires Java 8.
| option | description | default value |
|---|---|---|
| --in | Required list of input sam or bam file(s) separated by comma | - |
| --out | Required list of output sam or bam file(s) separated by comma | - |
| --ref | Genome reference location | - |
| --targets | BED file containing target regions | - |
| --target-kmers | BED-like file containing target regions with per region kmer sizes in 4th column | - |
| --kmer | Optional assembly kmer size(delimit with commas if multiple sizes specified) | - |
| --mnf | Assembly minimum node frequency | 1 |
| --mcl | Assembly minimum contig length | -1 |
| --threads | Number of threads | 4 |
| --single | Input is single end | - |
| --mapq | Minimum mapping quality for a read to be used in assembly and be eligible for realignment | 20 |
| --rcf | Minimum read candidate fraction for triggering assembly | 0.01 |
| --mad | Regions with average depth exceeding this value will be downsampled | 1000 |
| --mer | Min edge pruning ratio. Default value is appropriate for relatively sensitive somatic cases. May be increased for improved speed in germline only cases. | 0.01 |
| --maxn | Maximum pre-pruned nodes in regional assembly | 150000 |
| --sa | Skip assembly | - |
| --ssc | Skip usage of soft clipped sequences as putative contigs | - |
| --sc | Soft clip contig args [max_contigs,min_base_qual,frac_high_qual_bases,min_soft_clip_len] | 16,13,80,15 |
| --sobs | Do not use observed indels in original alignments to generate contigs | - |
| --junctions | Splice junctions definition file | - |
| --log | Logging level (trace,debug,info,warn,error) | info |
| --contigs | Optional file to which assembled contigs are written | - |
| --gtf | GTF file defining exons and transcripts | - |
| --sga | Scoring used for contig alignments (match, mismatch_penalty, gap_open_penalty, gap_extend_penalty) | 8,32,48,1 |
| --mcr | Max number of cached reads per sample per thread | 1000000 |
| --keep-tmp | Do not delete the temporary directory | - |
| --tmpdir | Set the temp directory (overrides java.io.tmpdir) | - |
| --cons | Use positional consensus sequence when aligning high quality soft clipping | - |
| --mmr | Max allowed mismatch rate when mapping reads back to contigs | 0 |
| --ws | Processing window size and overlap (size,overlap) | 400,200 |
| --mrr | Regions containing more reads than this value are not processed. Use -1 to disable. | 1000000 |
| --cl | Compression level of output bam file(s) | 5 |
| --ca | Contig anchor [M_bases_at_contig_edge, max_mismatches_near_edge] | 10,2 |
| --nosort | Do not attempt to sort final output | - |
| --dist | Max read move distance | 1000 |
| --skip | If no target specified, skip realignment of chromosomes matching specified regex. Skipped reads are output without modification. Specify none to disable. | GL.* hs37d5 chr.*random chrUn.* chrEBV CMV HBV HCV.* HIV.* KSHV HTLV.* MCV SV40 HPV.* |
| --mac | Max assembled contigs | 64 |
| --sua | Do not use unmapped reads anchored by mate to trigger assembly. These reads are still eligible to contribute to assembly | - |
| --undup | Unset duplicate flag | - |
| --in-vcf | VCF containing known (or suspected) variant sites. Very large files should be avoided. | - |
| --index | Enable BAM index generation when outputting sorted alignments (may require additonal memory) | - |
| --gkl | If specified, use GKL Intel Deflater (experimental) | - |
| --amq | Set mapq for alignments that map equally well to reference and an ABRA generated contig. default of -1 disables | -1 |
| --mrn | Reads with noise score exceeding this value are not remapped. numMismatches+(numIndels2) < readLengthmnr | .10 |
| --msr | Max reads to keep in memory per sample during the sort phase. When this value is exceeded, sort spills to disk | 1000000 |
Sample command for DNA:
java -Xmx16G -jar abra2.jar --in normal.bam,tumor.bam --out normal.abra.bam,tumor.abra.bam --ref hg38.fa --threads 8 --targets targets.bed --dist 1000 --tmpdir /your/tmpdir > abra.log
The above accepts normal.bam and tumor.bam as input and outputs sorted realigned BAM files named normal.abra.bam and tumor.abra.bam
- Input files must be sorted by coordinate and index
- Output files are sorted
- The tmpdir may grow large. Be sure you have sufficient space there (at least equal to the input file size)
- The targets argument is not required. When omitted, the entire genome will be eligible for realignment.
-
the picture is the original bam,we can see many reads have softclip in the end
-
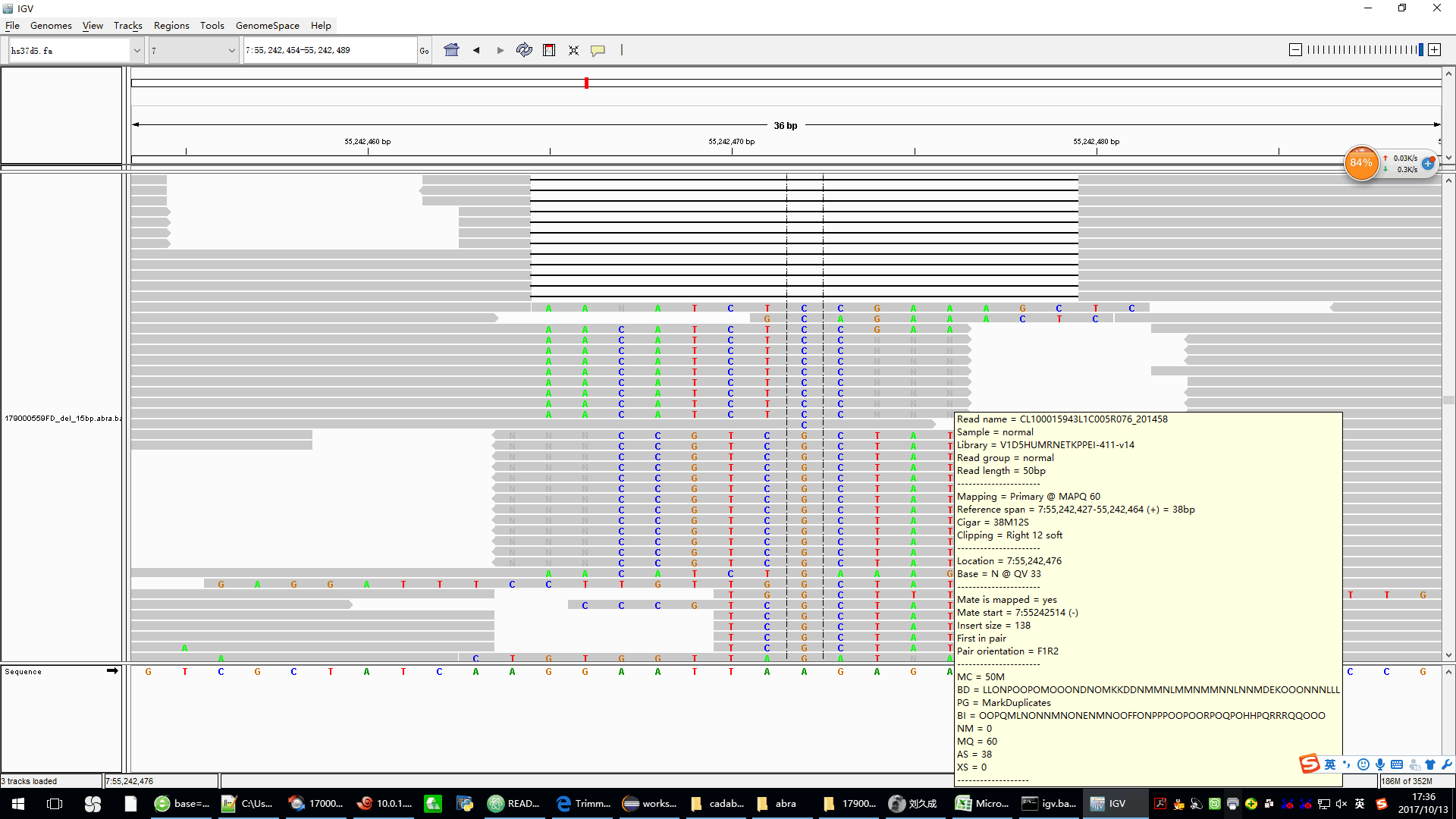

* the picture is the abra2 processed bam,we can see some reads have softclip in the end,these reads contains N in the end

* the picture is the abra2_modified processed bam,we can see only a few reads have softclip in the end,these reads and their mate reads are not mapped in the same chromosome

Abra2 is capable of utilizing junction information to aid in assembly and realignment. It has been tested only on STAR output to date.
Sample command for RNA:
java -Xmx16G -jar abra2.jar --in star.bam --out star.abra.bam --ref hg38.fa --junctions SJ.out.tab --threads 8 --gtf gencode.v26.annotation.gtf --dist 500000 --tmpdir /your/tmpdir > abra2.log 2>&1
Here, star.bam is the input bam file and star.abra.bam is the output bam file.
Junctions observed during alignment can be passed in using the --junctions param. This corresponds to the SJ.out.tab file output by STAR.
Annotated junctions can be passed in using the --gtf param. See: https://www.gencodegenes.org/releases/current.html
It is beneficial to use both of the junction related options.
The software is currently considered beta quality.