The goal of this project is to enable you to utilize genomic big data in identifying regulatory mechanisms for differential expression (DE).
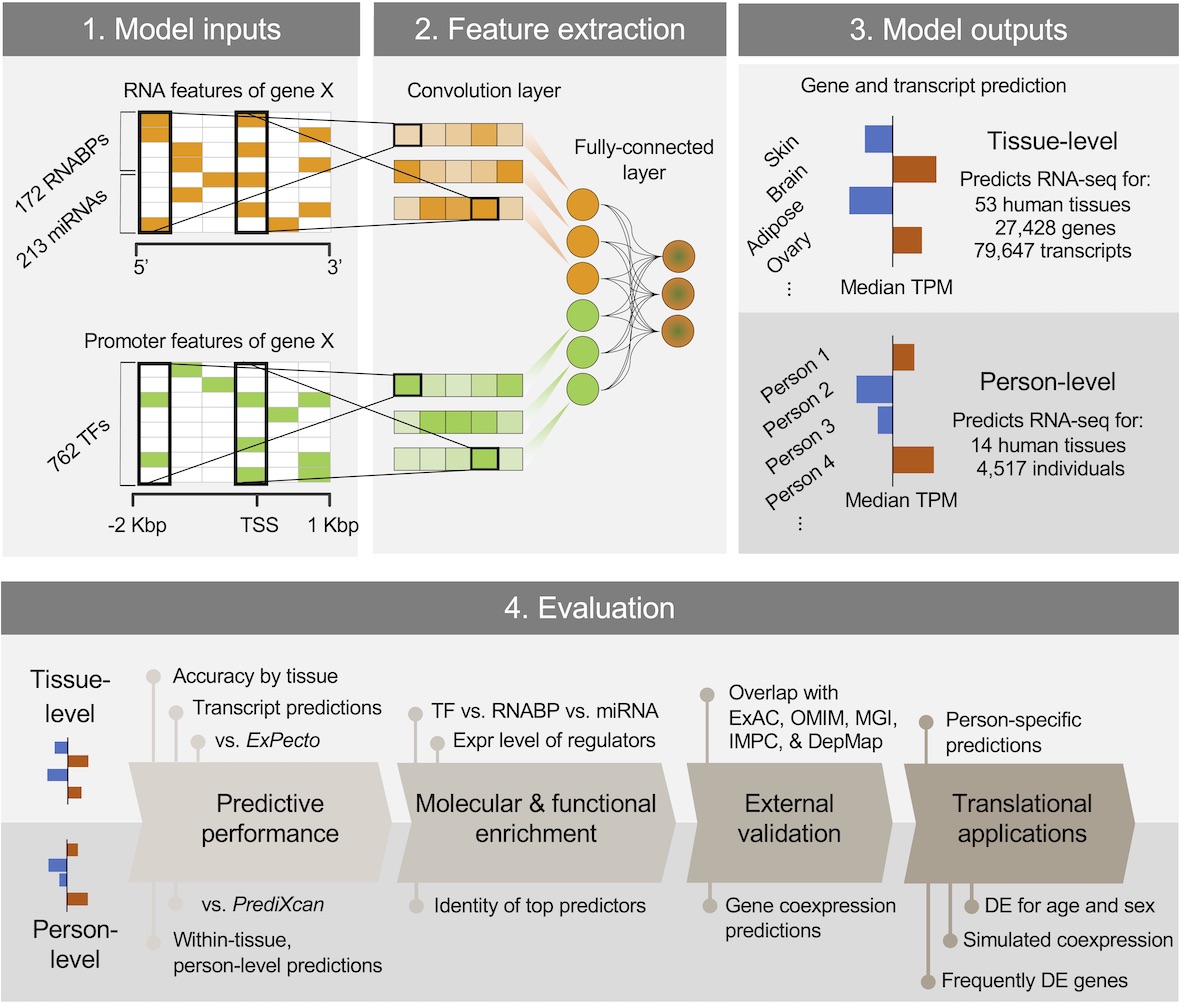
DEcode predicts inter-tissue variations and inter-person variations in gene expression levels from TF-promoter interactions, RNABP-mRNA interactions, and miRNA-mRNA interactions.
You can read more about this method in this paper (full text is available at https://rdcu.be/b5r3p) where we conducted a series of evaluation and applications by predicting transcript usage, drivers of aging DE, gene coexpression relationships on a genome-wide scale, and frequent DE in diverse conditions.
You can run DEcode on Code Ocean platform without setting up a computational environment. Our Code Ocean capsule provides reproducible workflows, all processed data, and pre-trained models for tissue- and person-specific transcriptomes and DEprior, at gene- or transcript level.
Before running the scripts to create custom input data, you must have the following python libraries installed on your system:
- pandas
- scipy
Also you need to install the following R packages:
- GenomicFeatures
- tidyverse
- data.table
- rtracklayer
- plyranges
- optparse
- First, download the example files by running the following commands:
#GTF file from GTEXv7 (hg19)
mkdir gtf
wget https://storage.googleapis.com/gtex_analysis_v7/reference/gencode.v19.genes.v7.patched_contigs.gtf -P ./gtf/
#eCLIP-seq peaks from Encode (hg19)
mkdir bed_rna
wget https://www.encodeproject.org/files/ENCFF039BKT/@@download/ENCFF039BKT.bed.gz -P ./bed_rna/
wget https://www.encodeproject.org/files/ENCFF379UQU/@@download/ENCFF379UQU.bed.gz -P ./bed_rna/
#ChIP-seq peaks from Encode (hg19)
mkdir bed_promoter
wget https://www.encodeproject.org/files/ENCFF553GPK/@@download/ENCFF553GPK.bed.gz -P ./bed_promoter/
wget https://www.encodeproject.org/files/ENCFF549TYR/@@download/ENCFF549TYR.bed.gz -P ./bed_promoter/This will create directories gtf, bed_rna, and bed_promoter, and download the GTF file, two eCLIP-seq bed files, and two ChIP-seq bed files into them.
Our input data for the gene-level model was constructed based on the gencode.v19.transcripts.patched_contigs.gtf file from the GTEXv7 dataset. This file contains only one representative transcript for each gene for the human genome (hg19).
If you use a different gene model, please make sure to filter GTF file and select a representative transcript for each gene before running the pipeline.
The input data for the transcript-level model was created based on https://storage.googleapis.com/gtex_analysis_v7/reference/gencode.v19.transcripts.patched_contigs.gtf. It is not necessary to filter the GTF file for the transcript-level model.
- Convert genome coordinates to RNA coordinates using the following command:
Rscript functions/bed_to_RNA_coord.R -b ./bed_rna/ -n 100 -g gtf/gencode.v19.genes.v7.patched_contigs.gtf -t rna -o custom_RNA- bed_directory: Character string specifying the directory containing the bed files
- bin: Numeric value specifying the size of bins for the genomic features
- gtf_file: Character string specifying the path to the GTF file
- input_type: Character string specifying the experiment type of the bed files, i.e. "promoter" or "rna"
- output: Character string specifying the path and filename of the output file
This will convert bed files in the genome coordinates in the ./bed_rna/ directory to RNA coordinates using the gencode.v19.genes.v7.patched_contigs.gtf file and output as custom_RNA.txt.
If you want to map ChIP-seq peaks to promoters, use the -t option as promoter.
Rscript functions/bed_to_RNA_coord.R -b ./bed_promoter/ -n 100 -g gtf/gencode.v19.genes.v7.patched_contigs.gtf -t promoter -o custom_promoter- To convert RNA-coordinate peaks to Pandas format, use the following command:
python functions/to_sparse.py custom_RNA.txt
python functions/to_sparse.py custom_promoter.txt
# clean up
rm custom_RNA.txt
rm custom_promoter.txt This will convert the RNA-coordinate peaks in the custom_RNA.txt file and the custom_promoter.txt file to sparse Pandas DataFrames (custom_RNA.pkl and custom_promoter.pkl).
-
Place
custom_RNA.pkl,custom_RNA_gene_name.txt.gz, andcustom_RNA_feature_name.txt.gzin the directory where RNA features are located, for example:./data/toy/RNA_features/. Also placecustom_promoter.pkl,custom_promoter_gene_name.txt.gz, andcustom_promoter_feature_name.txt.gzin the directory for promoter features, for example:./data/toy/Promoter_features/. -
Modify the code (Run_DEcode_toy.ipynb) as follows:
mRNA_data_loc = "./data/toy/RNA_features/"
mRNA_annotation_data = ["POSTAR","TargetScan","custom_RNA"]
promoter_data_loc = "./data/toy/Promoter_features/"
promoter_annotation_data = ["GTRD","custom_promoter"]This modification to the code will instruct it to utilize the custom_RNA.pkl and custom_promoter.pkl files as part of the RNA annotation data and promoter annotation data, respectively.
Tasaki, S., Gaiteri, C., Mostafavi, S. & Wang, Y. Deep learning decodes the principles of differential gene expression. Nature Machine Intelligence (2020) [link to paper] (full text is available at https://rdcu.be/b5r3p)
- GTEx transcriptome data - GTEx portal
- Transcription factor binding peaks - GTRD
- RNA binding protein binding peaks - POSTAR2
- miRNA binding locations - TargetScan