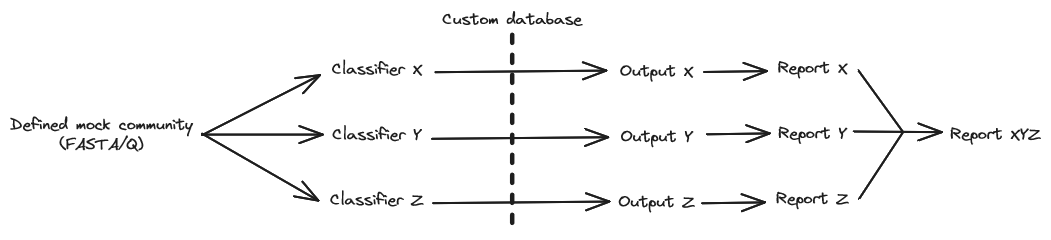
BenchmarkClassifiers is a framework designed to evaluate the performance of various taxonomic classifiers given defined mock communities (DMC). Following the classification of each DMC, a standardized output is produced of every classifier. Subsequently, this output is formatted into a report containing tables and figures.
git clone https://github.com/BioinformaticsPlatformWIV-ISP/BenchmarkingClassifiers.git
biopython==1.81
click==8.1.7
Jinja2==3.1.2
matplotlib==3.8.2
numpy==1.26.2
pandas==2.1.3
PyYAML==6.0.1
requests==2.31.0
rich==13.7.0
seaborn==0.13.0
scikit-learn==1.3.2
snakemake==7.32.4
pip3 install pulp==2.7.0
seqkit/2.3.1
taxonkit/0.15.0
python3 run_map_classifiers.py -h
usage: run_map_classifiers.py [-h] --input INPUT --truth TRUTH --output OUTPUT --config_file CONFIG_FILE [--classifier [...]] [--dir_working DIR_WORKING] [--dry_run] [--no_filter]
options:
-h, --help show this help message and exit
--input INPUT FASTA/Q input file
--truth TRUTH Location to the ground truth file.
--output OUTPUT Output of snakemake
--config_file CONFIG_FILE
YAML file with location of tools and database.
Template can be found under "/snakemake/config/classifiers.yml.template"
--classifier [ ...] Which classifier(s) to use.
Default choices are ['bracken', 'ccmetagen', 'centrifuge', 'kaiju', 'kma', 'kraken2', 'metaphlan', 'mmseqs2', 'motus']; space separated.
If not set, all default choices will be used.
--dir_working DIR_WORKING
Working directory.
If not set, it will be {output path} + "snakemake_conf".
--dry_run Run the SnakeMake file in Dry Run mode
--no_filter Disable filtering of reads (length > 1000 and quality > 7)Each classifier will generate two output files, one for the genus level and one for the species level. Such an output file consists of two tab-separated columns. The first column contains the organism's name, while the second column indicates the number of reads (or the taxonomic relative abundance for marker-based classifiers) assigned to that particular organism.
A SnakeMake workflow was made for the following classifiers:
| Tool | Version | Dependancies |
|------------|-----------|-----------------------------|
| bracken | 2.8 | |
| ccmetagen | 1.4.1 | |
| centrifuge | 1.0.4 | |
| kaiju | 1.9.0 | |
| kraken2 | 2.1.1 | |
| kma | 1.3.28 | |
| metaphlan | 3.0.14 | bowtie2==2.5.1 |
| MMseqs2 | 13.45111 | |
| motus | 2.6.0 | bwa==0.7.17; samtools==1.17 |
Paths to tools, databases and dependencies are stored in the snakemake/config/classifiers.yml file.
The REQUIRED section contains an entry for the taxonomy database. This folder should contain the uncompressed taxdump of NCBI.
The CLASSIFIERS should contain the paths to the executable and database for classifiers. If a classifier is a Python module, the path should indicate the environment where the classifier is installed.
The DEPENDENCIES section should contain the path to potential dependencies.
The taxonomic tree of NCBI undergoes continuous changes. As a result, if built on different dates, the ground truth and the output of classifiers may refer to the same organism but have different taxids. The workflow ensures consistency by updating both the ground truth and the output of classifiers to the same NCBI tadump's version specified in the config file. An updated ground truth file named ground_truth_updated.yml will be written out to the --output directory.
python3 generate_report.py -h
usage: generate_report.py [-h] --input INPUT --output OUTPUT [--classifier CLASSIFIER [CLASSIFIER ...]]
options:
-h, --help show this help message and exit
--input INPUT Output from SnakeMake flow. Format is [name]/ouput/reads.
--output OUTPUT Location of report/figures
--classifier CLASSIFIER [CLASSIFIER ...]
Which classifier(s) to use [all]python3 generate_report_aggregated.py -h
usage: generate_report_aggregated.py [-h] -i -o
options:
-h, --help show this help message and exit
-i , --input Output from SnakeMake flow
-o , --output Location of report/figuresA subsampled dataset of ERR2906227 can be found in the repo under the folderexample along with the ground truth.
The dataset can be classified using the command:
python3 run_map_classifiers.py --input /ERR2906227_subsampled.fq --truth ground_truth.yml --output example/zymo_d6300 --config classifiers.yml --classifier kma, kraken2The output folder is organised as:
zymo_d6300
├── input <-- contains the filtered input dataset (length > 1000 and Q > 7)
├── kma <-- contains output files of KMA
├── kraken2 <-- contains output files of Kraken2
├── logs <-- log files of the pipeline
├── output <-- processed output of all classifier on genus/species level
│ ├── genus
│ │ ├── output_kma.tsv
│ │ └── output_kraken2.tsv
│ └── species
│ ├── output_kma.tsv
│ └── output_kraken2.tsv
└── snakemake_conf <-- SnakeMake config filesThe output_*.tsv output files contain the species names and corresponding number of reads (or relative abundance for taxonomic classifiers) per line:
output_kma.tsv
Bacillus spizizenii 16
Limosilactobacillus fermentum 12
Listeria monocytogenes 12
Enterococcus faecalis 10
Pseudomonas aeruginosa 4
Salmonella enterica 4
Escherichia coli 3
Saccharomyces cerevisiae 3
Staphylococcus aureus 2
Shigella flexneri 1
Cryptococcus neoformans 1
no_hit 3The report can be generated with:
python3 generate_report.py --input example/zymo_d6300/output/ --output example/zymo_d6300/report/The generated report can be found under example
An aggregated report can be generated with:
python3 generate_report_aggregated.py --input example/ --output report_agg/