This is a python class for dealing with VASP pseudo-wavefunction file WAVECAR.
It can be used to extract the planewave coefficients of any single Kohn-Sham (KS)
orbital from the file. In addition, by padding the planewave coefficients to a
3D grid and performing 3D Fourier Transform, the pseudo-wavefunction in real
space can also be obtained and saved to file that can be viewed with VESTA.
With the knowledge of the planewave coefficients of the pseudo-wavefunction, transition dipole moment between any two KS states can also be calculated.
IPR is a measure of the localization of Kohn-Sham states. For a particular KS state \phi_j, it is defined as
\sum_n |\phi_j(n)|^4
IPR(\phi_j) = -------------------------
|\sum_n |\phi_j(n)|^2||^2where n iters over the number of grid points.
(Still need to be tested!)
In quantum chemistry, the electron localization function (ELF) is a measure of the likelihood of finding an electron in the neighborhood space of a reference electron located at a given point and with the same spin. Physically, this measures the extent of spatial localization of the reference electron and provides a method for the mapping of electron pair probability in multielectronic systems. (from wiki)
- Nature, 371, 683-686 (1994)
- Becke and Edgecombe, J. Chem. Phys., 92, 5397(1990)
- M. Kohout and A. Savin, Int. J. Quantum Chem., 60, 875-882(1996)
- http://www2.cpfs.mpg.de/ELF/index.php?content=06interpr.txt
NOTE that if you are using VESTA to view the resulting ELF file, please rename the output file as "ELFCAR", otherwise there will be some error in the isosurface plot! When VESTA read in CHG*/PARCHG/*.vasp to visualize isosurfaces and sections, data values are divided by volume in the unit of bohr^3. The unit of charge densities input by VESTA is, therefore, bohr^−3. For LOCPOT/ELFCAR files, volume data are kept intact.
Using the pseudo-wavefunction from supercell calculation, it is possible to perform electronic band structure unfolding to obtain the effective band structure. For more information, please refer to the following article and the GPAW website.
V. Popescu and A. Zunger Extracting E versus k effective band structure from supercell calculations on alloys and impurities Phys. Rev. B 85, 085201 (2012)
-
Manual Installation
Put
vasp_constant.pyandvaspwfc.pyin any directory you like and add the path of the directory toPYTHONPATHexport PYTHONPATH=/the/path/of/your/dir:${PYTHONPATH}
requirements
- numpy
- scipy
- matplotlib
-
Using Pip
pip install git+https://github.com/QijingZheng/VaspBandUnfolding
- Write a simple script and choose whichever state you like.
from vaspwfc import vaspwfc
wav = vaspwfc('./examples/wfc_r/WAVECAR')
# KS orbital in real space, double the size of the FT grid
phi = wav.wfc_r(ikpt=2, iband=27, ngrid=wav._ngrid * 2)
# Save the orbital into files. Since the wavefunction consist of complex
# numbers, the real and imaginary part are saved separately.
wav.save2vesta(phi, poscar='./examples/wfc_r/POSCAR')
# for WAVECAR from a noncollinear run, the wavefunction at each k-piont/band is
# a two component spinor. Turn on the lsorbit flag when reading WAVECAr.
xx = vaspwfc('examples/wfc_r/wavecar_mose2-wse2', lsorbit=True)
phi_spinor = xx.wfc_r(1, 1, 36, ngrid=xx._ngrid*2)
for ii in range(2):
phi = phi_spinor[ii]
prefix = 'spinor_{:02d}'.format(ii)
xx.save2vesta(phi, prefix=prefix,
poscar='examples/wfc_r/poscar_mose2-wse2')Below are the real (left) and imaginary (right) part of the selected KS orbital:
 |
|

- Or you can also use the script
wfcplotin thescriptsfolder
$ wfcplot -w WAVECAR -p POSCAR -s spin_index -k kpoint_index -n band_index # for normal WAVECAR
$ wfcplot -w WAVECAR -p POSCAR -s spin_index -k kpoint_index -n band_index -lgamma # for gamma-only WAVECAR
$ wfcplot -w WAVECAR -p POSCAR -s spin_index -k kpoint_index -n band_index -lgamma # for noncollinear WAVECARPlease refer to wfcplot -h for more information of the usage.
import numpy as np
from vaspwfc import vaspwfc, save2vesta
kptw = [1, 6, 6, 6, 6, 6, 6, 12, 12, 12, 6, 6, 12, 12, 6, 6]
wfc = vaspwfc('./WAVECAR')
# chi = wfc.elf(kptw=kptw, ngrid=wfc._ngrid * 2)
chi = wfc.elf(kptw=kptw, ngrid=[20, 20, 150])
save2vesta(chi[0], lreal=True, poscar='POSCAR', prefix='elf')Remember to rename the output file "elf_r.vasp" as "ELFCAR"!
Here, we use MoS2 as an example to illustrate the procedures of band unfolding. Another example can be found on my website.
Below is the band structure of MoS2 using a primitive cell. The calculation was
performed with VASP and the input files can be found in the
examples/unfold/primitive

-
Create the supercell from the primitive cell, in my case, the supercell is of the size 3x3x1, which means that the transformation matrix between supercell and primitive cell is
# The tranformation matrix between supercell and primitive cell. M = [[3.0, 0.0, 0.0], [0.0, 3.0, 0.0], [0.0, 0.0, 1.0]]
-
In the second step, generate band path in the primitive Brillouin Zone (PBZ) and find the correspondig K points of the supercell BZ (SBZ) onto which they fold.
from unfold import make_kpath, removeDuplicateKpoints, find_K_from_k # high-symmetry point of a Hexagonal BZ in fractional coordinate kpts = [[0.0, 0.5, 0.0], # M [0.0, 0.0, 0.0], # G [1./3, 1./3, 0.0], # K [0.0, 0.5, 0.0]] # M # create band path from the high-symmetry points, 30 points inbetween each pair # of high-symmetry points kpath = make_kpath(kpts, nseg=30) K_in_sup = [] for kk in kpath: kg, g = find_K_from_k(kk, M) K_in_sup.append(kg) # remove the duplicate K-points reducedK, kid = removeDuplicateKpoints(K_in_sup, return_map=True) # save to VASP KPOINTS save2VaspKPOINTS(reducedK)
-
Do one non-SCF calculation of the supercell using the folded K-points and obtain the corresponding pseudo-wavefunction. The input files are in
examples/unfold/sup_3x3x1/. The effective band structure (EBS) and then be obtained by processing the WAVECAR file.from unfold import unfold # basis vector of the primitive cell cell = [[ 3.1850, 0.0000000000000000, 0.0], [-1.5925, 2.7582909110534373, 0.0], [ 0.0000, 0.0000000000000000, 35.0]] WaveSuper = unfold(M=M, wavecar='WAVECAR') from unfold import EBS_scatter sw = WaveSuper.spectral_weight(kpath) # show the effective band structure with scatter EBS_scatter(kpath, cell, sw, nseg=30, eref=-4.01, ylim=(-3, 4), factor=5) from unfold import EBS_cmaps e0, sf = WaveSuper.spectral_function(nedos=4000) # or show the effective band structure with colormap EBS_cmaps(kpath, cell, e0, sf, nseg=30, eref=-4.01, show=False, ylim=(-3, 4))
The EBS from a 3x3x1 supercell calculation are shown below:
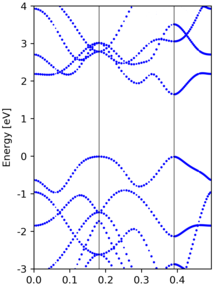 |
|
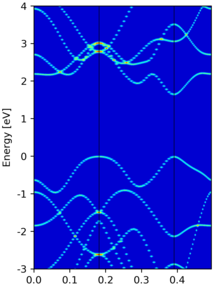
Another example of EBS from a 3x3x1 supercell calculation, where we introduce a
Svacancy in the structure.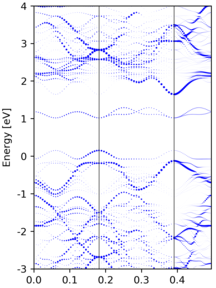 |
|
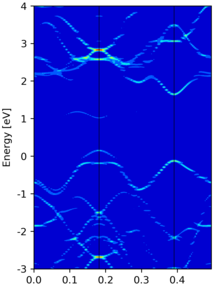
Yet another band unfolding example from a tetragonal 3x3x1 supercell calculation, where the transformation matrix is
M = [[3.0, 0.0, 0.0], [3.0, 6.0, 0.0], [0.0, 0.0, 1.0]]
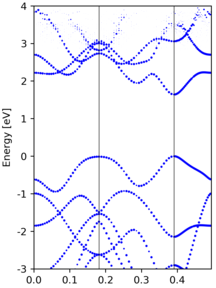 |
|
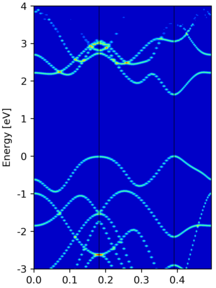
Compared to the band structure of the primitive cell, there are some empty states at the top of figure. This is due to a too small value of
NBANDSin supercell non-scf calculation, and thus those states are not included.






After band unfolding, we can also superimpose the atomic contribution of each KS
states on the spectral weight. Below is the resulting unfolded band structure of
Ce-doped bilayer-MoS2. Refer to
./examples/unfold/Ce@BL-MoS2_3x3x1/plt_unf.py for the entire code.

Band re-ordering is possible by maximizing the overlap between nerghbouring k-points. The overlap is defined as the inner product of the periodic part of the Bloch wavefunctions.
`< u(n, k) | u(m, k-1) >`
Note, however, the WAVECAR only contains the pseudo-wavefunction, and thus the
pseudo u(n,k) are used in this function. Moreover, since the number of
planewaves for each k-points are different, the inner product is performed in
real space.
The overlap maximalization procedure is as follows:
- Pick out those bands with large overlap (> olap_cut).
- Assign those un-picked bands by maximizing the overlap.
An example band structure re-ordering is performed in MoS2. The result is shown in the following image, where the left/right panel shows the un-ordered/re-ordered band structure.
 |
|