The Metagenomics Evaluation & Testing Analysis (META) Tool is a software system that executes one or more third-party metagenomics classifiers on simulated nucleotide read sequences, and comparatively evaluates classification accuracy and runtime performance. In addition, the user can run a real-world read sequence (FASTQ) through any of the available classifiers view the aggregate results from one or more third-party metagenomics classifiers.
These instructions will get you a copy of the project up and running on your local machine for development and testing purposes.
The META system has been designed to run on Linux (specifically, tested on Ubuntu 18.04) and in Docker containers. The following packages are required:
Here is an example of how to install these on Ubuntu 18.04:
# Install Make
sudo apt-get update
sudo apt-get install build-essential
dpkg -l | grep automake # to verify successful install
# Install Docker engine (reference: https://docs.docker.com/engine/install/ubuntu/)
sudo apt-get remove docker docker-engine docker.io containerd runc
sudo apt-get update
sudo apt-get install \
apt-transport-https \
ca-certificates \
curl \
gnupg-agent \
software-properties-common
curl -fsSL https://download.docker.com/linux/ubuntu/gpg | sudo apt-key add -
sudo add-apt-repository \
"deb [arch=amd64] https://download.docker.com/linux/ubuntu \
$(lsb_release -cs) \
stable"
sudo apt-get update
sudo apt-get install docker-ce docker-ce-cli containerd.io
sudo docker run hello-world # to verify successful install
# Install Docker-compose (reference: https://docs.docker.com/compose/install/)
sudo curl -L "https://github.com/docker/compose/releases/download/1.26.2/docker-compose-$(uname -s)-$(uname -m)" -o /usr/local/bin/docker-compose
sudo chmod +x /usr/local/bin/docker-compose
docker-compose --version # to verify successful installPrior to starting up META, you need to update your configurations for the:
- Locations of your metagenomic reference databases (default:
"/srv/databases") - MongoDB database port (default:
27017) - META server host bind (default:
"127.0.0.1") and port (default:5000) - Docker-in-Docker DNS Server for internet access (default:
"--dns 8.8.8.8")
To change these configuratons, go to the docker-compose.yaml file and update the following:
# Configure location of metagenomic reference databases
services:
# ...
meta_docker:
# ...
volumes:
- ./data:/data
- <INSERT_PATH_HERE>:/srv/databases # only modify left-hand side where <INSERT_PATH_HERE>
- meta_docker_data:/var/lib/docker
- meta_docker_certs_ca:/certs/ca
- meta_docker_certs_client:/certs/client
# ...
command: "--dns 8.8.8.8" # modify IP addresses to match your enterprise's DNS servers
# ...
meta_system:
volumes:
- ./data:/data
- <INSERT_PATH_HERE>:/srv/databases # only modify left-hand side where <INSERT_PATH_HERE>
- meta_docker_certs_client:/certs/client:ro# Configure MongoDB database port
services:
mongodb:
# ...
ports:
- "127.0.0.1:<INSERT_PORT_HERE>:27017" # only modify left-hand side where <INSERT_PORT_HERE># Configure META server host bind and port
services:
# ...
meta_system:
# ...
ports:
- "<INSERT_SERVER_HOST_BIND_HERE>:<INSERT_SERVER_PORT_HERE>:5000" # only modify left-hand side where <INSERT_SERVER_HOST_BIND_HERE> and <INSERT_SERVER_PORT_HERE>If you do not have the META Simulator Docker image on your machine, you will need to do the following steps:
- Clone META Simulator from https://github.com/JHUAPL/meta-simulator
- From the
meta-simulatordirectory, run:docker build -t meta_simulator:latest . docker save -o meta_simulator.tar meta_simulator:latest - Copy the
meta_simulator.tartometa_system/data/docker - Run
make load-dockerfrom themeta_systemdirectory
META uses make to handle building and install META system.
To deploy using Docker, run:
make build-docker
make up-docker
make logs-docker # to verify successful install (see below)You can verify your application is running successfully by checking that the following logs are visible in your terminal window:
2020-07-23 19:19:17,976 shared.log INFO LOGGING INITIALIZED
2020-07-23 19:19:18,034 shared.log DEBUG STARTING JOB QUEUE WATCHDOG
* Serving Flask app "system" (lazy loading)
* Environment: production
WARNING: This is a development server. Do not use it in a production deployment.
Use a production WSGI server instead.
* Debug mode: on
* Running on http://0.0.0.0:5000/ (Press CTRL+C to quit)
* Restarting with stat
2020-07-23 19:19:18,606 shared.log INFO LOGGING INITIALIZED
2020-07-23 19:19:18,656 shared.log DEBUG STARTING JOB QUEUE WATCHDOG
* Debugger is active!
* Debugger PIN: 219-671-456
2020-07-23 19:19:19,036 shared.log INFO WAITING FOR NEW JOBS...To begin using META, navigate to http://localhost:5000. You should see the following page in your web browser:

In effect, there are two job modes that META can run:
- Simulate + Classify: This feature allows you to initiate a job pipeline that simulates sequencing from the Oxford Nanopore R9 flowcell (
r9), Oxford Nanopore Flongle flowcell (flg), Illumina iSeq (iseq), or Illumina MiSeq (miseq) platforms. These simulations generate a FASTQ file that approximate a given abundance profile. For each simulated FASTQ, a set of user-selected classifiers return a predicted abundance profile that is then compared to the user-specific abundance profile. The list of currently available classifiers is shown in the table below.- An abundance profile is expressed as a tab-delimited text file (TSV) where the first column contains the leaf taxonomic ID, the second column contains the corresponding abundance proportion (must sum to 1.000000), and the third column designates the organism as being foreground (
1) or background (0). There should be no headers in the abundance profile TSV. An example is shown below:400667 0.10 1 435590 0.10 1 367928 0.10 1 864803 0.10 1 1091045 0.10 1 349101 0.10 1 1282 0.10 1 260799 0.10 1 1529886 0.10 1 198094 0.10 1
- An abundance profile is expressed as a tab-delimited text file (TSV) where the first column contains the leaf taxonomic ID, the second column contains the corresponding abundance proportion (must sum to 1.000000), and the third column designates the organism as being foreground (
- Classify: This feature allows you to initiate a job pipeline that accepts a FASTQ file and runs a set of user-selected classifiers. The list of currently available classifiers is shown below. The metagenomic classifiers currently integrated in META include:
| Tool | Description | Average Runtime (seconds) for 1.0 GB FASTQ File |
|---|---|---|
| Kraken | k-mer based, exact alignment | 4.95 |
| Kraken2 | k-mer based, exact alignment, with smaller memory requirements, and translated search mode | 0.32 |
| KrakenUniq | k-mer based, exact alignment with smaller memory requirements | 71.05 |
| Bracken | k-mer based abundance estimation from raw reads | 0.04 |
| Centrifuge | alignment based on BWT and FM-indexing schemes | 14.38 |
| DIAMOND | alignment based, SW against protein database | 145.47 |
| Mash | k-mer based, locality sensitive hashing | 0.64 |
| MMseqs2 | k-mer based | 0.07 |
| BLASTN/P | alignment based, basic local alignment | 74.08 |
| Kaiju | alignment based on BWT and FM-indexing schemes using protein sequences | 14.63 |
| Kallisto | alignment based, pseudoalignment procedure | 0.05 |
| CLARK | k-mer based species or genus level classification | 0.04 |
It is possible to add new classifiers to META by following the instructions in ADD_BIOCONTAINERS.md.
- Click the Submit Jobs button in the Navigation Bar.
- Configure the job with
- the Job Title
- the mode you would like to use Simulate + Classify or Classify
- upload an abundance profile TSV file or a FASTQ file
- A sample abundance profile is included in
meta_system/data/sample_abundance_profile.tsv- To start Simulate + Classify jobs in bulk, you may submit a
.zipfolder of abundance profiles (i.e. a zipped folder of.tsvfiles). You will be provided the option to Upload ZIP after selecting Simulate + Classify. This will create a new row in the View Jobs table for each.tsvfile contained in the ZIP folder. It is recommended that the TSV files in the folder are uniquely named.
- To start Simulate + Classify jobs in bulk, you may submit a
- A sample FASTQ file is included in
meta_system/data/fastq/sample.fastq- To start Classify jobs in bulk, you may submit a
.zipfolder of abundance profiles (i.e. a zipped folder of.fastqfiles). You will be provided the option to Upload ZIP after selecting Classify. This will create a new row in the View Jobs table for each.fastqfile contained in the ZIP folder. It is recommended that the FASTQ files in the folder are uniquely named.
- To start Classify jobs in bulk, you may submit a
- A sample abundance profile is included in
- the classifiers you would like to use
- the simulation read type, if you selected the Simulated + Classify mode
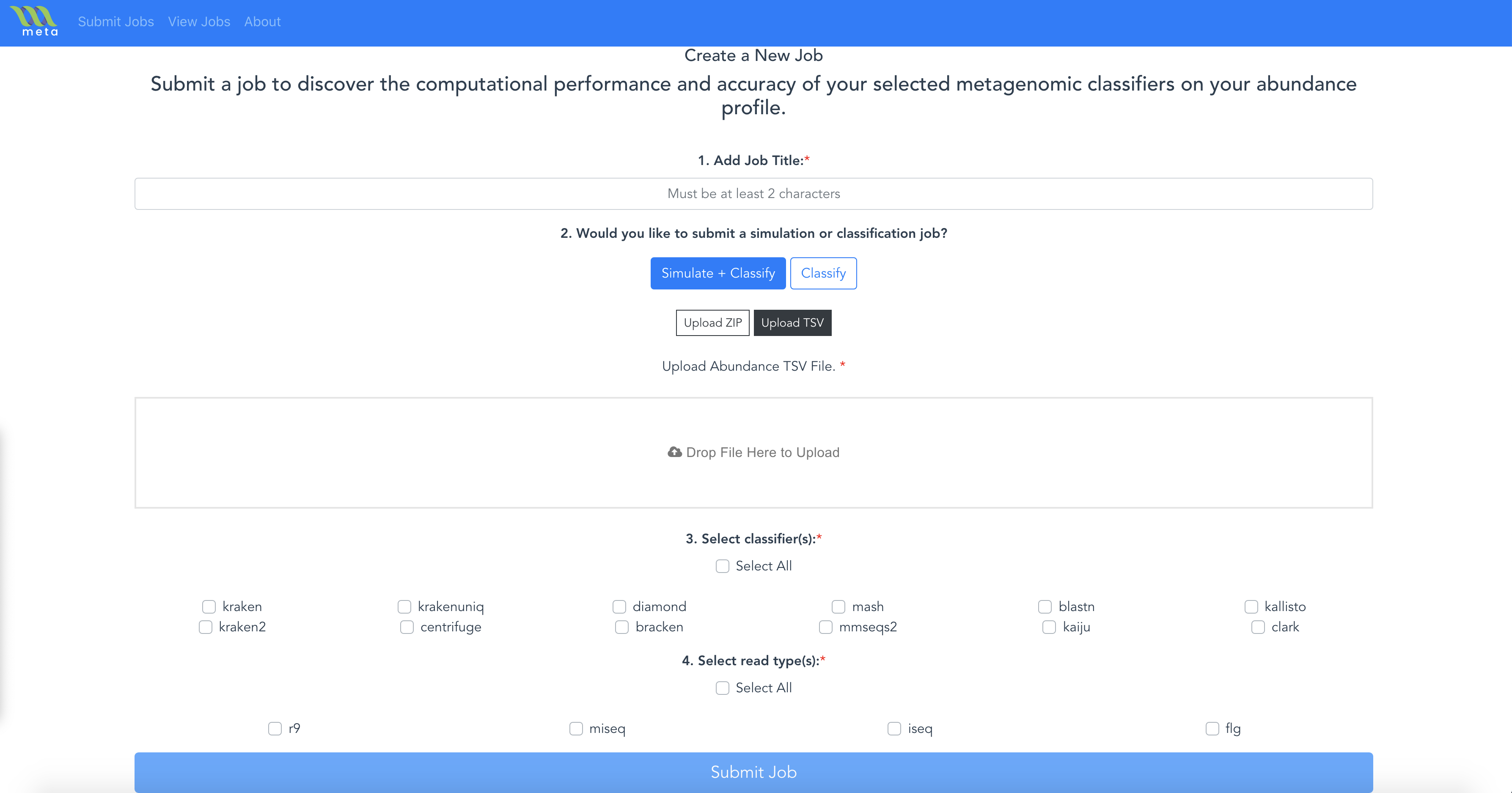
- Click Submit Job

If you would like to install a development version (outside of Docker), the following additional packages are required:
Here is an example of how to install these on Ubuntu 18.04:
# Install Parallel
sudo apt-get install parallel
dpkg -l | grep parallel # to verify successful install
# Install Python 3.7+ (reference: https://linuxize.com/post/how-to-install-python-3-7-on-ubuntu-18-04/)
sudo apt update
sudo apt install software-properties-common
sudo add-apt-repository ppa:deadsnakes/ppa
sudo apt install python3.7
python3 --version # to verify successful install
# Install Node 10+ (reference: https://joshtronic.com/2018/05/08/how-to-install-nodejs-10-on-ubuntu-1804-lts/)
sudo apt install curl
curl -sL https://deb.nodesource.com/setup_10.x | sudo -E bash -
sudo apt install nodejs
node --version # to verify successful install
# Install Node Package Manager (NPM)
sudo apt install npm
npm --version # to verify successful installPrior to starting up META, you need to update your configurations for the:
- META server host bind (default:
"0.0.0.0") and port (default:5000) - Locations of your metagenomic reference databases (default:
"") - MongoDB database port (default:
27018)
To change these configuratons, go to the meta_system/shared/config.py file and update the following:
class MetaConfiguration(BaseSettings):
# ...
# Modify META server host bind and port
SERVER_BIND: str = "0.0.0.0" # <-- Modify server host bind here
SERVER_PORT: str = 5000 # <-- Modify port number here
# ...
# Configure Biocontainers info and database paths
# ...
BIOCONTAINER_DB_DIR: str = "" # <--- Modify location of metagenomic reference databases
# ...
# Configure MongoDB
# ...
MONGO_PORT: int = 27017 # <-- Modify MongoDB port hereTo deploy a development server, run:
make build
make up-db
make upPlease read CONTRIBUTING.md for details on our code of conduct, and the process for submitting pull requests to us.
Here are some planned improvements, in no particular order:
- Add ability to compare between select job results
- Add prediction model to determine the runtime of a job prior to executing
- Add metagenomic assemblers
- Add configurable read counts for simulated reads
- Add configurable thread counts for metagenomic classifiers
- Add export capability for graphs and visualizations as PNG, JPEG, PDF, etc.
- Add ability to construct abundance profile for simulation within the user interface
- Add user management
We use SemVer for versioning. For the versions available, see the tags on this repository.
This project is licensed under Apache 2.0. Copyright under Johns Hopkins University Applied Physics Laboratory.