![]()
ProteinFlow - A data processing pipeline for all your protein design needs



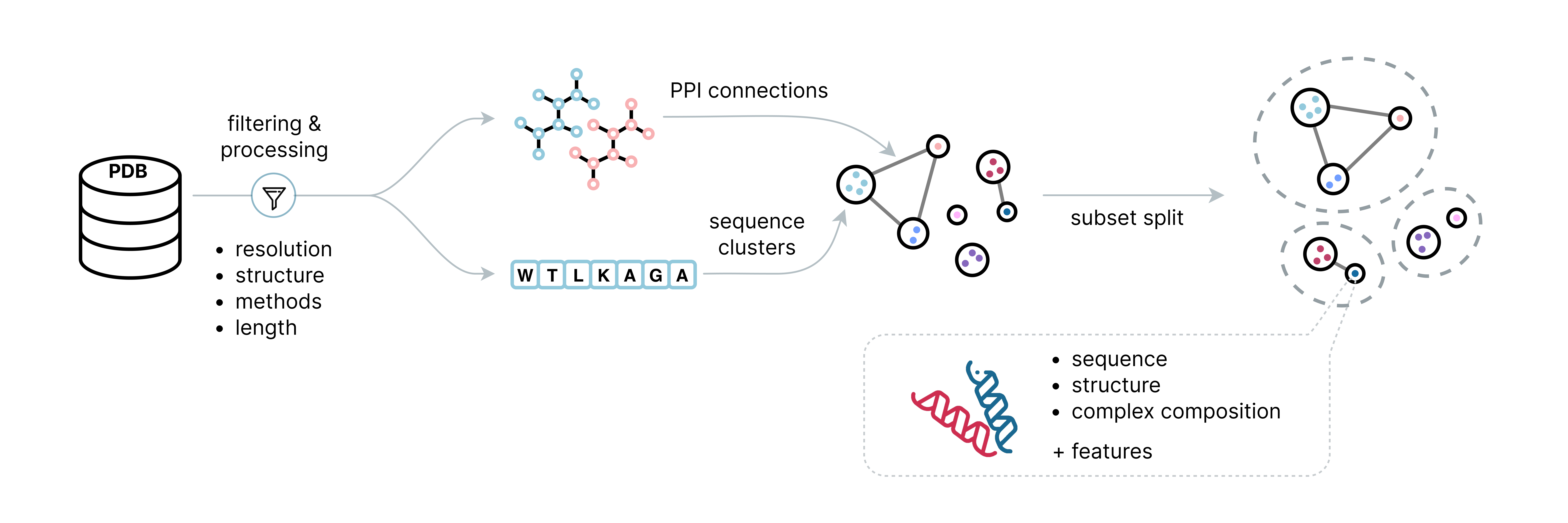
ProteinFlow is an open-source Python library that streamlines the pre-processing of protein structure data for deep learning applications. ProteinFlow enables users to efficiently filter, cluster, and generate new datasets from resources like the Protein Data Bank (PDB) and SAbDab (The Structural Antibody Database).
Here are some of the key features we currently support:
- ⛓️ Processing of both single-chain and multi-chain protein structures (Biounit PDB definition)
- 🏷️ Various featurization options can be computed, including secondary structure features, torsion angles, etc.
- 💾 A variety of data loading options and conversions to cater to different downstream training frameworks
- 🧬 Access to up-to-date, pre-computed protein structure datasets
conda:
# This should take a few minutes, be patient
conda install -c conda-forge -c bioconda -c adaptyvbio proteinflowpip:
pip install proteinflowdocker:
docker pull adaptyvbio/proteinflowBy default installing proteinflow with conda or pip will only load the dependencies that are required for the main functions of the package: downloading, generating and splitting datasets. If you are interested in using other functions like visualization, metrics and other data processing methods, please install the package with pip install proteinflow[processing] or use the docker image.
Some metric functions also have separate requirements, see the documentation for details. All of them are installed in the docker image.
- If you are using python 3.10 and encountering installation problems, try running
python -m pip install prody==2.4.0before installingproteinflow. - If you are planning to generate new datasets and installed
proteinflowwithpip(or withcondaon Mac OS with an M1 processor), you will need to additionally installmmseqs. - Generating new datasets also depends on the
rcsbsearchpackage and the latest release v0.2.3 is currently not working correctly. The recommended fix is installing the version from this pull request.
python -m pip install "rcsbsearch @ git+https://github.com/sbliven/rcsbsearch@dbdfe3880cc88b0ce57163987db613d579400c8e"- The docker image can be accessed in interactive mode with this command.
docker run -it -v /path/to/data:/media adaptyvbio/proteinflow bashAlready precomputed datasets with consensus set of parameters and can be accessed and downloaded using the proteinflow. package. Check the output of proteinflow check_tags for a list of available tags.
proteinflow download --tag 20230102_stable You can also run proteinflow with your own parameters. Check the output of proteinflow check_snapshots for a list of available PDB snapshots (naming rule: yyyymmdd).
For instance, let's generate a dataset with the following description:
- resolution threshold: 5 angstrom,
- PDB snapshot: 20190101,
- structure methods accepted: all (x-ray christolography, NRM, Cryo-EM),
- sequence identity threshold for clustering: 40% sequence similarity,
- maximum length per sequence: 1000 residues,
- minimum length per sequence: 5 residues,
- maximum fraction of missing values at the ends: 10%,
- size of validation subset: 10%.
proteinflow generate --tag new --resolution_thr 5 --pdb_snapshot 20190101 --not_filter_methods --min_seq_id 0.4 --max_length 1000 --min_length 5 --missing_ends_thr 0.1 --valid_split 0.1See the docs (or proteinflow generate --help) for the full list of parameters and more information.
A registry of all the files that are removed during the filtering as well as description with the reason for their removal is created automatically for each generate command. The log files are save (at data/logs by default) and a summary can be accessed running proteinflow get_summary {log_path}.
You can also use the --sabdab option in proteinflow generate to load files from SAbDab and cluster them based on CDRs. By default the --sabdab tag will download the latest up-to-date version of the SabDab dataset and cluster the antibodies based on their CDR sequence.
Alternatively, it can be used together with the tag --sabdab_data_path to process a custom SAbDab-like zip file or folder. This allows you to use search and query tools from the SabDab web interface to create a custom dataset by downloading the archived zip file of the structures selected. (Under Downloads section of your SabDab query).
SAbDab sequences clustering is done across all 6 Complementary Determining Regions (CDRs) - H1, H2, H3, L1, L2, L3, based on the Chothia numbering implemented by SabDab. CDRs from nanobodies and other synthetic constructs are clustered together with other heavy chain CDRs. The resulting CDR clusters are split into training, test and validation in a way that ensures that every PDB file only appears in one subset.
Individual output pickle files represent heavy chain - light chain - antigen complexes (created from SAbDab annotation, sometimes more than one per PDB entry). Each of the elements (heavy chain, light chain, antigen) can be missing in specific entries and there can be multiple antigen chains. In order to filter for at least one antigen chain, use the --require_antigen option.
For instance, let's generate a dataset with the following description:
- SabDab version: latest (up-to-date),
- resolution threshold: 5 angstrom,
- structure methods accepted: all (x-ray christolography, NRM, Cryo-EM),
- sequence identity threshold for clustering (CDRs): 40%,
- size of validation subset: 10%.
proteinflow generate --sabdab --tag new --resolution_thr 5 --not_filter_methods --min_seq_id 0.4 --valid_split 0.1By default, both proteinflow generate and proteinflow download will also split your data into training, test and validation according to MMseqs2 clustering and homomer/heteromer/single chain proportions. However, you can skip this step with a --skip_splitting flag and then perform it separately with the proteinflow split command.
The following command will perform the splitting with a 10% validation set, a 5% test set and a 50% threshold for sequence identity clusters.
proteinflow split --tag new --valid_split 0.1 --test_split 0.5 --min_seq_id 0.5Use the --exclude_chains and --exclude_threshold parameters to move all biounits that contain chains similar to what you specify to a separate folder.
The output files are pickled nested dictionaries where first-level keys are chain Ids and second-level keys are the following:
'crd_bb': anumpyarray of shape(L, 4, 3)with backbone atom coordinates (N, C, CA, O),'crd_sc': anumpyarray of shape(L, 10, 3)with sidechain atom coordinates (checkproteinflow.sidechain_order()for the order of atoms),'msk': anumpyarray of shape(L,)where ones correspond to residues with known coordinates and zeros to missing values,'seq': a string of lengthLwith residue types.
In a SAbDab datasets, an additional key is added to the dictionary:
'cdr': anumpyarray of shape(L,)where CDR residues are marked with the corresponding type ('H1','L1', ...) and non-CDR residues are marked with'-'.
Note that the sequence information in the PDB files is aligned to the FASTA sequences to identify the missing residues.
Once your data is ready, you can open the files with pickle directly.
import pickle
import os
train_folder = "./data/proteinflow_new/training"
for filename in os.listdir(train_folder):
with open(os.path.join(train_folder, filename), "rb") as f:
data = pickle.load(f)
crd_bb = data["crd_bb"]
seq = data["seq"]
...Alternatively, you can use our ProteinDataset or ProteinLoader classes
for convenient processing. Among other things, they allow for feature extraction, single chain / homomer / heteromer filtering and randomized sampling from sequence identity clusters.
For example, here is how we can create a data loader that:
- samples a different cluster representative at every epoch,
- extracts dihedral angles, sidechain orientation and secondary structure features,
- only loads pairs of interacting proteins (larger biounits are broken up into pairs),
- has batch size 8.
from proteinflow import ProteinLoader
train_loader = ProteinLoader.from_args(
"./data/proteinflow_new/training",
clustering_dict_path="./data/proteinflow_new/splits_dict/train.pickle",
node_features_type="dihedral+sidechain_orientation+secondary_structure",
entry_type="pair",
batch_size=8,
)
for batch in train_loader:
crd_bb = batch["X"] #(B, L, 4, 3)
seq = batch["S"] #(B, L)
sse = batch["secondary_structure"] #(B, L, 3)
to_predict = batch["masked_res"] #(B, L), 1 where the residues should be masked, 0 otherwise
...See more details on available parameters and the data format in the docs + this repository for a use case.
You can download them with proteinflow download --tag {tag} in the command line or browse in the interface.
| Tag | Date | Snapshot | Size | Min res | Min len | Max len | Max chains | MMseqs thr | Split (train/val/test) | Missing thr (ends/middle) | Source | Remove redundancies | Note |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| paper | 10.11.22 | 20220103 | 24G | 3.5 | 30 | 10'000 | - | 0.3 | 90/5/5 | 0.3/0.1 | PDB | yes | first release, no mmCIF files |
| 20230102_stable | 27.02.23 | 20230102 | 28G | 3.5 | 30 | 10'000 | - | 0.3 | 90/5/5 | 0.3/0.1 | PDB | yes | v1.1.1 |
| 20230623_sabdab | 26.06.23 | live 26.06.23 | 1.4G | 3.5 | 30 | 10'000 | - | 0.3 | 96/3/1 | 0.5/0.2 | SAbDab | no | v1.4.1 (requires >= v1.4.0) |
| 20230102_v200 | 19.07.23 | 20230102 | 33G | 3.5 | 30 | 10'000 | 10 | 0.3 | 90/5/5 | 0.3/0.1 | PDB | no | v2.0.0 |
| 20231221_sabdab | 21.12.23 | live 21.12.23 | 1.8G | 3.5 | 30 | 10'000 | - | 0.3 | 96/3/1 | 0.5/0.2 | SAbDab | no | v2.6.1 (requires >= v1.4.0) |
The proteinflow package and data are released and distributed under the BSD 3-Clause License
This is an open source project supported by Adaptyv Bio. Contributions, suggestions and bug-fixes are welcomed.