This repository is made for eBook entitled “Bioinformatics Recipes for Plant Genomics: Data, Code, and Workflows” in Bio-101
Hi-C is a chromosome conformation capture method that was originally developed to detect genome-wide chromatin interactions. Nowadays, it is widely applied in scaffolding de novo assembled contigs into chromosome-scale genome sequences. Multiple open-source software has been developed to perform genome scaffolding with Hi-C data. The input data is de novo assembled contigs using long-read sequencing or next-generation sequencing. Then, Hi-C data is mapped to these contigs and the interact matrix is computed by software to scaffold contigs into chromosome-scale sequences. Different tools have their specific algorithm to calculate the interaction matrix, correct misassembly and misjoins, and also may require different dependent packages or running environments. Here, we describe a step-by-step protocol for genome scaffolding using Hi-C data with a comprehensive pipeline: compute interact matrix with Juicer, scaffold contigs with 3D-DNA pipeline, then visualize and modify scaffolding with Juicebox. This pipeline only requires primarily assembled contigs and Hi-C data as inputs, is compatible with multiple enzymes, and also provides visualization and manual correction. With these advantages, this pipeline has been used in mass big eukaryotic genome scaffolding.
Trimmomatic
Juicer
3D-DNA pipeline
Juicebox
BWA
Samtools
BUSCO
Java 1.8 JDK
- De novo assembly contigs file in FASTA format, contigs used in this study can be downloaded HERE
- Raw Hi-C sequencing data in FASTQ format, fastq files used in this study can be downloaed HERE
More details can be found in the paper
a. Linux server or cluster with SLURM or SLF scheduler
b. PC with at least 16GB RAM for handling big genomes (>1GB)
mkdir hic; cd hic
git clone https://github.com/theaidenlab/juicer.git
ln -s juicer/SLURM/scripts/ scripts
cd scripts; wget https://hicfiles.tc4ga.com/public/juicer/juicer_tools.1.9.9_jcuda.0.8.jar
ln -s juicer_tools.1.9.9_jcuda.0.8.jar juicer_tools.jar; cd ../
mkdir references
mkdir restriction_sites
And make sure samtools and bwa are in your $PATH
a. Copy your_contigs.fasta file (or make soft link) into reference path, and index it with bwa index
ln -s your_path/your_contigs.fasta ./references
cd ./references; bwa index your_contig.fasta; cd ..
b. Prepare enzyme site file for your_contigs.fasta
cd restriction_sites
wget https://raw.githubusercontent.com/aidenlab/juicer/main/misc/generate_site_positions.py
Then, Use vi or vim to edit generate_site_positions.py, insert the following line in line 25:
'your_contigs': '../references/your_contigs.fasta',
After that:
python generate_site_positions.py DpnII your_contigs
awk 'BEGIN{OFS="\t"}{print $1, $NF}' your_contigs_DpnII.txt > your_contigs.chrom.sizes
cd ..
c. Filter and clean raw Hi-C sequencing data
wget https://github.com/usadellab/Trimmomatic/files/5854859/Trimmomatic-0.39.zip
unzip Trimmomatic-0.39.zip
java -jar ./Trimmomatic-0.39/trimmomatic-0.39.jar PE -threads 12 -phred33 -trimlog trimmomatic.log your_hic_R1.fastq.gz your_hic_R2.fastq.gz your_hic_pair_R1.fastq.gz your_hic_unpair_R1.fastq.gz your_hic_pair_R2.fastq.gz your_hic_unpair_R2.fastq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
mkdir your_contigs_hic; cd your_contigs_hic
mkdir fastq
ln -s ../your_hic_pair* ./fastq/
sh ../scripts/juicer.sh -D $pwd/hic -g your_contigs -s DpnII -p ../restriction_sites/your_contigs.chrom.sizes -y ../restriction_sites/your_contigs_DpnII.txt -z ../references/your_contig.fasta -Q 2-00:00 -L 7-00:00 -q your_queue_name -l your_long_queue_name -t 24 -A your_account --assembly
cd ..
Tips: Check juicer.sh, make sure all the Partition, Account, QOS and Threads fit your SLURM or SLF scheduler. To be safe, add these parameters to your command line or modify juicer.sh manually.
Juicer will submit jobs to the cluster through the scheduler automatically. After all the jobs are done, the file named merged_nodups.txt in your_contigs_hic/aligned will be used by 3D-DNA pipeline.
wget https://github.com/aidenlab/3d-dna/archive/refs/tags/201008.tar.gz
tar -zxf 201008.tar.gz
chmod 554 ./3d-dna-201008/*.sh ##add execute permission
cd your_contigs_hic
../3d-dna-201008/run-asm-pipeline.sh ../references/your_contigs.fasta ./aligned/merged_nodups.txt
Tips: If the scaffolding results are not ideal, try different --round different edit round and slightly increase --editor-repeat-coverage misjoin editor threshold repeat coverage. In this case study, we use --editor-repeat-coverage 3. After the job is done. Two files named your_contigs.rawchrom.assembly and your_contigs.rawchrom.hic will be used by Juicebox
a. Download Juicebox v1.11.08 from https://github.com/aidenlab/Juicebox/wiki/Download
b. Download your_contigs.FINAL.assembly and your_contigs.rawchrom.hic to your PC
c. Run Juicebox, then load your_contigs.rawchrom.hic and your_contigs.FINAL.assembly in turn (Figure 1)
 Figure 1. Steps to load .hic and .assembly file to Juicebox. A. Load .hic file; B. Load .assembly file.
Figure 1. Steps to load .hic and .assembly file to Juicebox. A. Load .hic file; B. Load .assembly file.
d. Correct scaffolding manually (Figure 2 and Figure 3)
- Shift+left-click to choose the region that needs to be edited.
- Right-click to choose to remove or add chr boundaries.
- Move the mouse to the upper-right of the selected region until a circle appears, and then left-click to rotate the selected region
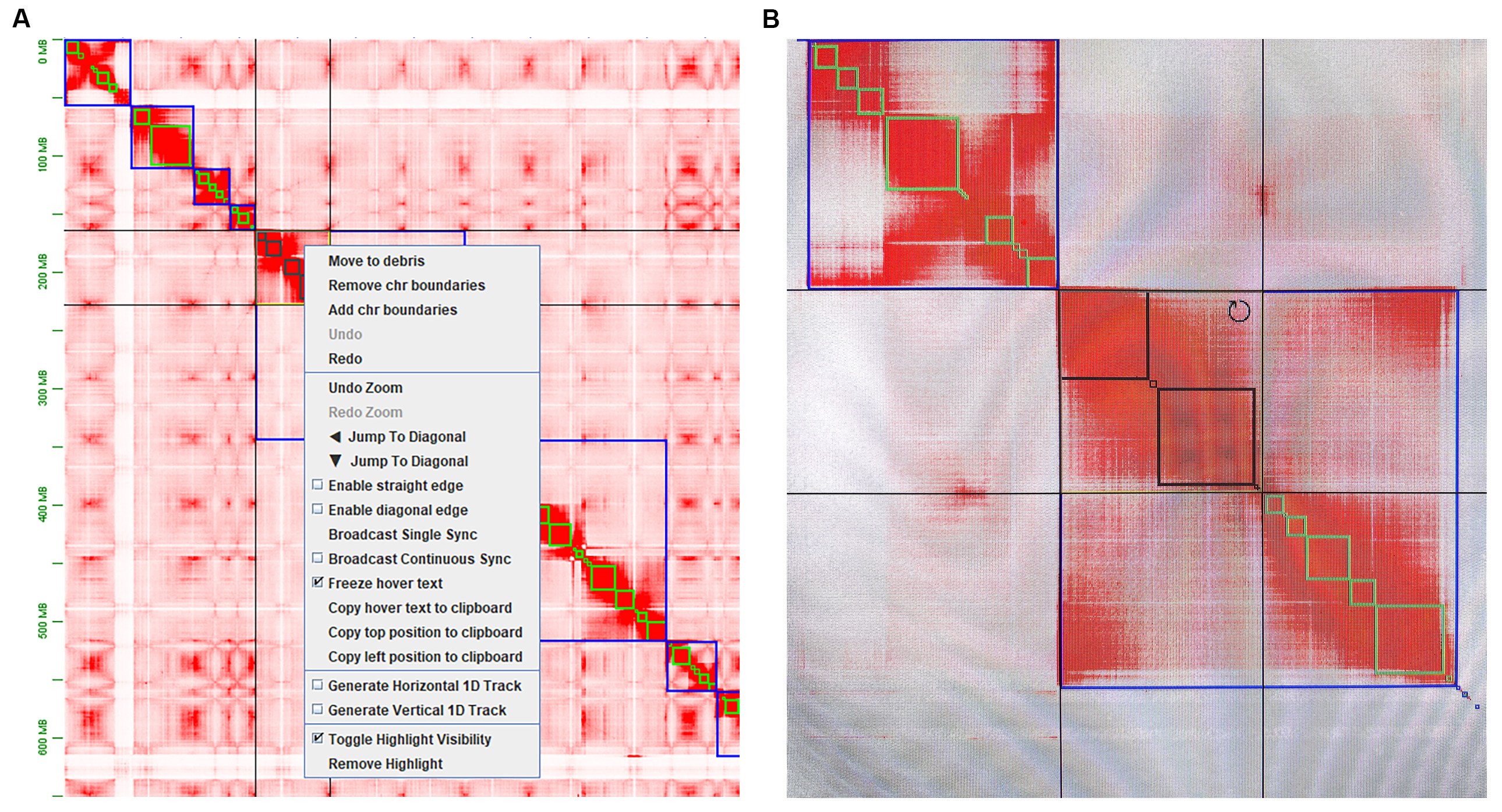 Figure 2. Examples of how to edit misjoin and misorientation. A. edit misjoin via remove and add chr boundaries. B. edit misorientation by rotating selected contigs.
Figure 2. Examples of how to edit misjoin and misorientation. A. edit misjoin via remove and add chr boundaries. B. edit misorientation by rotating selected contigs.
 Figure 3. Manually correct the scaffolding with Juicebox. A. the original scaffolding visilization B. manually corrected scaffolding. Ex. 1: misjoin, Ex. 2: mis-oritention
Figure 3. Manually correct the scaffolding with Juicebox. A. the original scaffolding visilization B. manually corrected scaffolding. Ex. 1: misjoin, Ex. 2: mis-oritention
Tips: Here is a demo video show how to use Juicebox from the software developer.


a. Upload your_contigs.FINAL.edit.assembly to cluster and put it in your_contigs_hic. b. Run 3D-DNA pipeline to obtain your edited scaffolds fasta file
../3d-dna/run-asm-pipeline-post-review.sh -r your_contigs.FINAL.edit.assembly ../references/your_contigs.fasta aligned/merged_nodups.txt
a. Install BUSCO, and download database
conda install -c bioconda busco
mkdir busco_evalue; cd busco_evalue
wget https://busco-data.ezlab.org/v4/data/lineages/embryophyta_odb10.2020-09-10.tar.gz
tar -zxf embryophyta_odb10.2020-09-10.tar.gz
b. Run BUSCO
busco -c 14 -m genome -i ../your_contigs_hic/your_contigs_HiC.fasta -o your_contigs_hic_busco -l ./embryophyta_odb10
c. Check the BUSCO value and components (Figure 4)
 Figure 4. BUSCO summary information.
Figure 4. BUSCO summary information.
It is a free and open source software, licensed under GPLv3