Kipoi: Model zoo for genomics


This repository implements a python package and a command-line interface (CLI) to access and use models from Kipoi-compatible model zoo's.
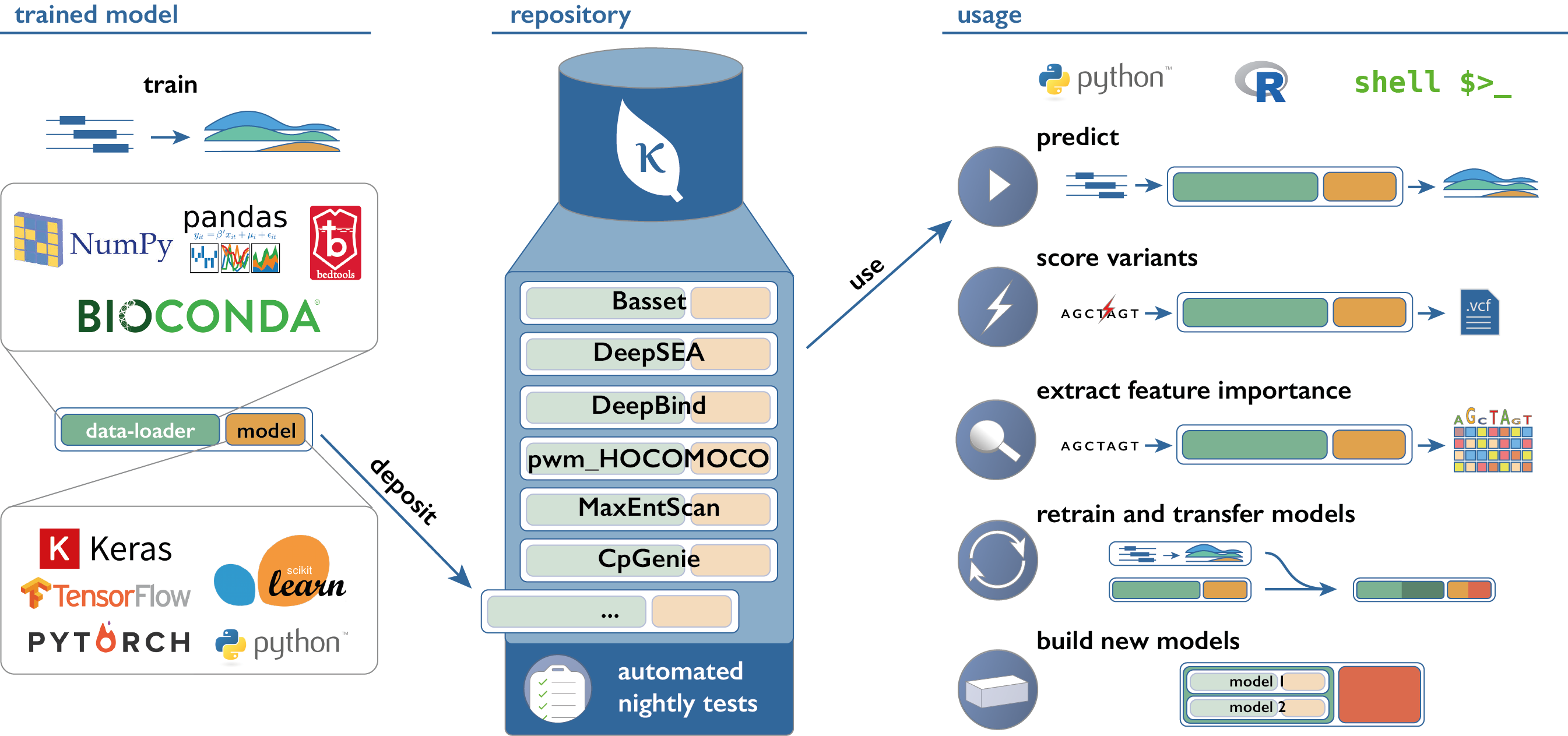
Links
- kipoi.org - Main website
- kipoi.org/docs - Documentation
- github.com/kipoi/models - Model zoo for genomics maintained by the Kipoi team
- biorxiv preprint - Kipoi: accelerating the community exchange and reuse of predictive models for genomics
- blog post introducing Kipoi, blog post describing 0.6 release
- github.com/kipoi/examples - Use-case oriented tutorials
Installation
Kipoi requires conda to manage model dependencies. Make sure you have either anaconda (download page) or miniconda (download page) installed. If you are using OSX, see Installing python on OSX. Supported python versions: 2.7 and >=3.5.
Install Kipoi using pip:
pip install kipoiQuick start
Explore available models on https://kipoi.org/groups/. Use-case oriented tutorials are available at https://github.com/kipoi/examples.
Installing all required model dependencies
Use kipoi env create <model> to create a new conda environment for the model. You can use the following two commands to create common environments suitable for multiple models.
kipoi env create shared/envs/kipoi-py3-keras2 # add --gpu to install gpu-compatible deps
kipoi env create shared/envs/kipoi-py3-keras1.2
Before using a model in any way, activate the right conda enviroment:
source activate $(kipoi env get <model>)Alternatively, you can use the Singularity or Docker containers with all dependencies installed. Singularity containers can be seamlessly used with the CLI by adding the --singularity flag to kipoi commands.
Python
import kipoi
kipoi.list_models() # list available models
model = kipoi.get_model("Basset") # load the model
model = kipoi.get_model( # load the model from a past commit
"https://github.com/kipoi/models/tree/<commit>/<model>",
source='github-permalink'
)
# main attributes
model.model # wrapped model (say keras.models.Model)
model.default_dataloader # dataloader
model.info # description, authors, paper link, ...
# main methods
model.dependencies.install() # calls `conda install` and `pip install`
model.predict_on_batch(x) # implemented by all the models regardless of the framework
model.pipeline.predict(dict(fasta_file="hg19.fa",
intervals_file="intervals.bed"))
# runs: raw files -[dataloader]-> numpy arrays -[model]-> predictions For more information see: notebooks/python-api.ipynb and docs/using/python
Command-line
$ kipoi
usage: kipoi <command> [-h] ...
# Kipoi model-zoo command line tool. Available sub-commands:
# - using models:
ls List all the available models
list_plugins List all the available plugins
info Print dataloader keyword argument info
get-example Download example files
predict Run the model prediction
pull Download the directory associated with the model
preproc Run the dataloader and save the results to an hdf5 array
env Tools for managing Kipoi conda environments
# - contributing models:
init Initialize a new Kipoi model
test Runs a set of unit-tests for the model
test-source Runs a set of unit-tests for many/all models in a source
# - plugin commands:
veff Variant effect prediction
interpret Model interpretation using feature importance scores like ISM, grad*input or DeepLIFT
# Run model predictions and save the results
# sequentially into an HDF5 file
kipoi predict <Model> --dataloader_args='{
"intervals_file": "intervals.bed",
"fasta_file": "hg38.fa"}' \
--singularity \
-o '<Model>.preds.h5'Explore the CLI usage by running kipoi <command> -h. Also, see docs/using/cli/ for more information.
Configure Kipoi in .kipoi/config.yaml
You can add your own (private) model sources. See docs/using/03_Model_sources/.
Contributing models
See docs/contributing getting started and docs/tutorials/contributing/models for more information.
Plugins
Kipoi supports plug-ins which are published as additional python packages. Two plug-ins that are available are:
kipoi_veff
Variant effect prediction plugin compatible with (DNA) sequence based models. It allows to annotate a vcf file using model predictions for the reference and alternative alleles. The output is written to a new VCF file. For more information see https://kipoi.org/veff-docs/.
pip install kipoi_veffkipoi_interpret
Model interpretation plugin for Kipoi. Allows to use feature importance scores like in-silico mutagenesis (ISM), saliency maps or DeepLift with a wide range of Kipoi models. example notebook
pip install kipoi_interpretDocumentation
Tutorials
- https://github.com/kipoi/examples - Use-case oriented tutorials
- notebooks
Citing Kipoi
If you use Kipoi for your research, please cite the publication of the model you are using (see model's cite_as entry) and our Biorxiv preprint: https://doi.org/10.1101/375345.
@article {kipoi,
author = {Avsec, Ziga and Kreuzhuber, Roman and Israeli, Johnny and Xu, Nancy and Cheng, Jun and Shrikumar, Avanti and Banerjee, Abhimanyu and Kim, Daniel S and Urban, Lara and Kundaje, Anshul and Stegle, Oliver and Gagneur, Julien},
title = {Kipoi: accelerating the community exchange and reuse of predictive models for genomics},
year = {2018},
doi = {10.1101/375345},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2018/07/24/375345},
eprint = {https://www.biorxiv.org/content/early/2018/07/24/375345.full.pdf},
journal = {bioRxiv}
}Development
If you want to help with the development of Kipoi, you are more than welcome to join in!
For the local setup for development, you should install kipoi using:
conda install pytorch-cpu
pip install -e '.[develop]'This will install some additional packages like pytest. You can test the package by running py.test.
If you wish to run tests in parallel, run py.test -n 6.