Different cell types and their associated functionalities can emerge from a single genomic sequence when certain regions are expressed while others remain silenced. The study of gene regulation and its potential malfunctioning in different cellular contexts is hence pivotal to understand both development and disease. We present the Chromatin Landscape and Structure to Expression Regressor (CLASTER), an epigenetic-based deep neural network that can integrate different data modalities describing the chromatin landscape and its 3D structure in their raw format. CLASTER effectively translates them into nascent transcription levels measured by EU-seq at a kilobasepair resolution. Our predictions reached a Pearson correlation with targets above r=0.86 at both bin and gene levels, without relying on DNA sequence nor explicitly extracted chromatin features. The model mostly used the information found within 10 kbp of the predicted locus to perform the predictions, even when a wide genomic region of 1 Mbp was available. Explicit modeling of long-range interactions using multi-headed attention and high-resolution chromatin contact maps had little impact on model performance, despite the model correctly identifying elements in these inputs influencing nascent transcription. The trained model then served as a platform to predict the transcriptional impact of simulated epigenetic silencing perturbations. Our results point towards a rather local, integrative and combinatorial paradigm of gene regulation, where changes in the chromatin environment surrounding a gene shape its context-specific transcription. We conclude that the predominant locality and limitations of current machine learning approaches might emerge as a genuine signature of genomic organization, having broad implications for future modeling approaches.
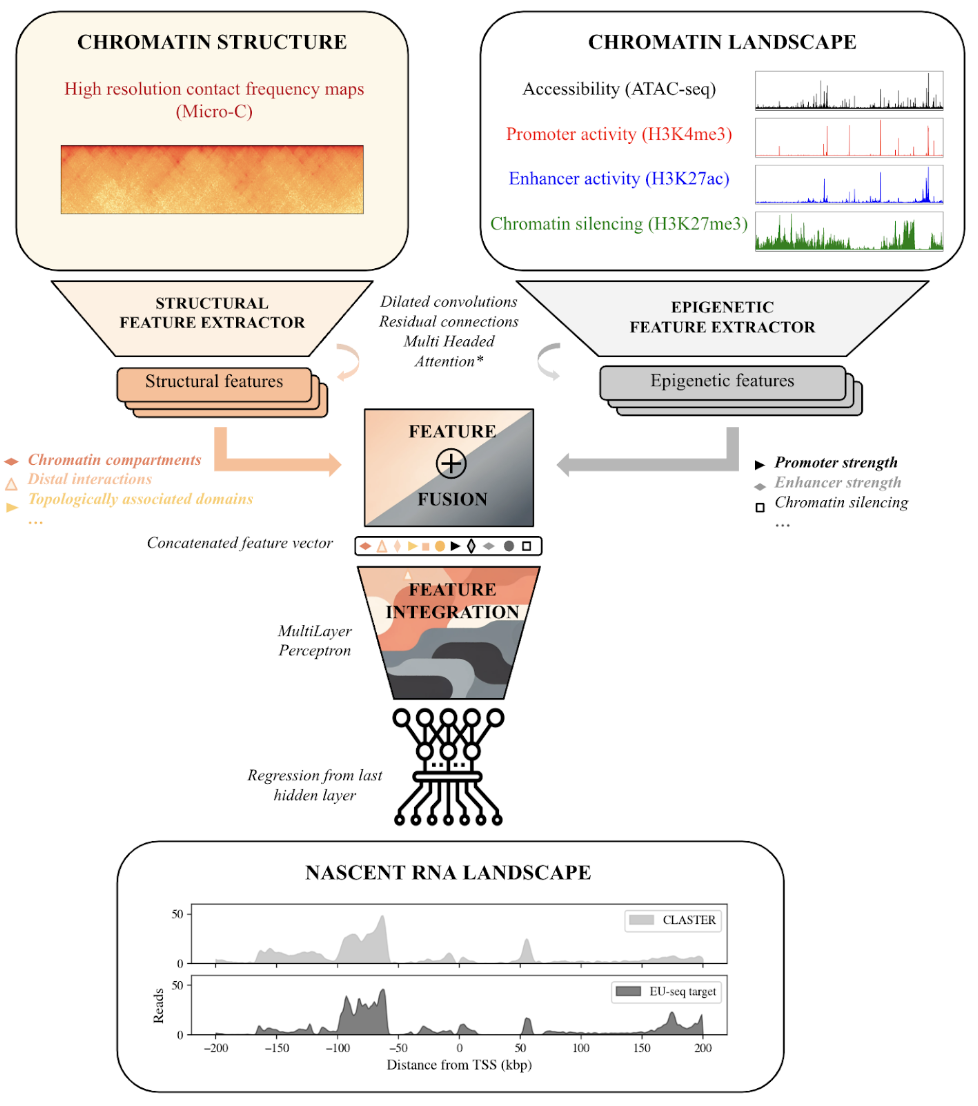
CLASTER overview CLASTER integrates the chromatin landscape (accessibility, promoter and enhancer activities and chromatin silencing) and structure (Micro-C) to predict nascent transcription levels measured by EU-seq.
This repository contains the files and scripts required to reproduce the results of the paper and a short tutorial. The repository consists of the following folders:
- Configuration files (.yaml) required to build different flavours of CLASTER.
- Overview of CLASTER's architecture.
The folder contains the test set inputs for both data modalities, i.e. samples exploring regions of 1 Mbp centered at the TSS of protein coding genes found in chr4 (in mice). They will be used in the tutorial to exemplify how can we train and validate CLASTER.
0_Tutorial.ipynb: The notebook provides a rapid overview of the most important steps in CLASTER's pipeline, including training and validating the network using the EIR framework.1_Data_obtention.ipynb: This notebook guides the user through the data obtention process, including:- Data download from publicly available repositories:
- Inputs: Chromatin landscape (ATAC-seq, H3K4me3, H3K27ac and H3K27me3 in mESCs) and structure (Micro-C maps in mESCs)
- Outputs: Nascent transcription profiles (EU-seq).
- Reference genome and gene annotations.
- Enhancer-Like Signatures (ELS).
- Data filtering and preprocessing:
- Obtain numpy arrays for the inputs.
- Obtain csv files for the targets.
- Data download from publicly available repositories:
2_Run_CLASTER.ipynb: This notebook creates the configuration files required to train and test CLASTER using the EIR framework.2b_Run_HyenaDNA_and_Enformer.ipynb: The notebook contains our adaptations of the code building- Hyena-DNA (https://github.com/HazyResearch/hyena-dna) in its public colab version.
- Enformer (https://github.com/lucidrains/enformer-pytorch) in its python implementation. These were used to benchmark CLASTER. It includes:
- The obtention of sequence embeddings from both model's backbones when loading the pretrained weights.
- The addition of a model head on top of the embeddings to match our regression outputs.
- Code to fine-tune Hyena-DNA's backbone and the added head together.
3_Data_analysis.ipynb: The notebook contains the functions used to perform the data analysis and create the figures included in the manuscript.
The folder contains the target EU-seq profiles matching the input (test) samples.