This repository contains the source code for the paper:
This pipeline is for processing multispectral fluorescence 2D image datasets. It will correct the multiplexed images for:
- Pixel-to-pixel registration due to stage misalignemnt between rounds
- Intra-channel non-specific signal correction of autofluorescence, non-uniform illumianation shading, Photo-pleaching and tissue folds
- Inter-channel non-specific signal correction of spectral bleed-through and molecular co-localization
And generate quantitative readouts for each cell including:
- Cell nuclei location
- Cell type
- Cell status
The pipeline is supported for Windows and Linux with CUDA-enabled GPU and enough RAM depending on the dataset size.
- numpy
- scipy
- pandas
- cython
- requests
- progressbar
- scikit-learn
- scikit-image==0.16.1
- tifffile
- opencv-python
- tensorflow-gpu==1.9.0
- pycocotools
- keras == 2.2.0
- h5py
setup_env.py creates a conda environment (brain) and installs the required libraries.
python setup_env.pyNote: tensorflow-gpu library requires CUDA toolkit 9.0 and cuDNN 7.0.5. You can follow the instructions from here to install the TensorFlow and dependencies.
For detection module you need to install protoc. Download executable from here and run the following command from
DETECTION/lib directory (read more):
# from DETECTION/lib
protoc object_detection/protos/*.proto --python_out=.Setting up the environment and installing the requirements takes ~45 minutes.
Parse the arguments to main_reconstruction.py:
python main_reconstruction.py \
--INPUT_DIR /path/to/input/dir \
--OUTPUT_DIR /path/to/output/dir \
--MODE supervised \
--SCRIPT /path/to/script.csvpython main_reconstruction.py \
--INPUT_DIR /path/to/input/dir \
--OUTPUT_DIR /path/to/output/dir \
--MODE unsupervised \
--DEFAULT_BOX 34000 8000 44000 15000 \
--BRIGHTFIELD 11Arguments:
-
INPUT_DIR: Path to the directory containing input images -
OUTPUT_DIR: Path to the directory saving output images -
MODE:supervisedorunsupervised -
if
unsupervised:DEFAULT_BOX:[xmin, ymin, xmax, ymax]
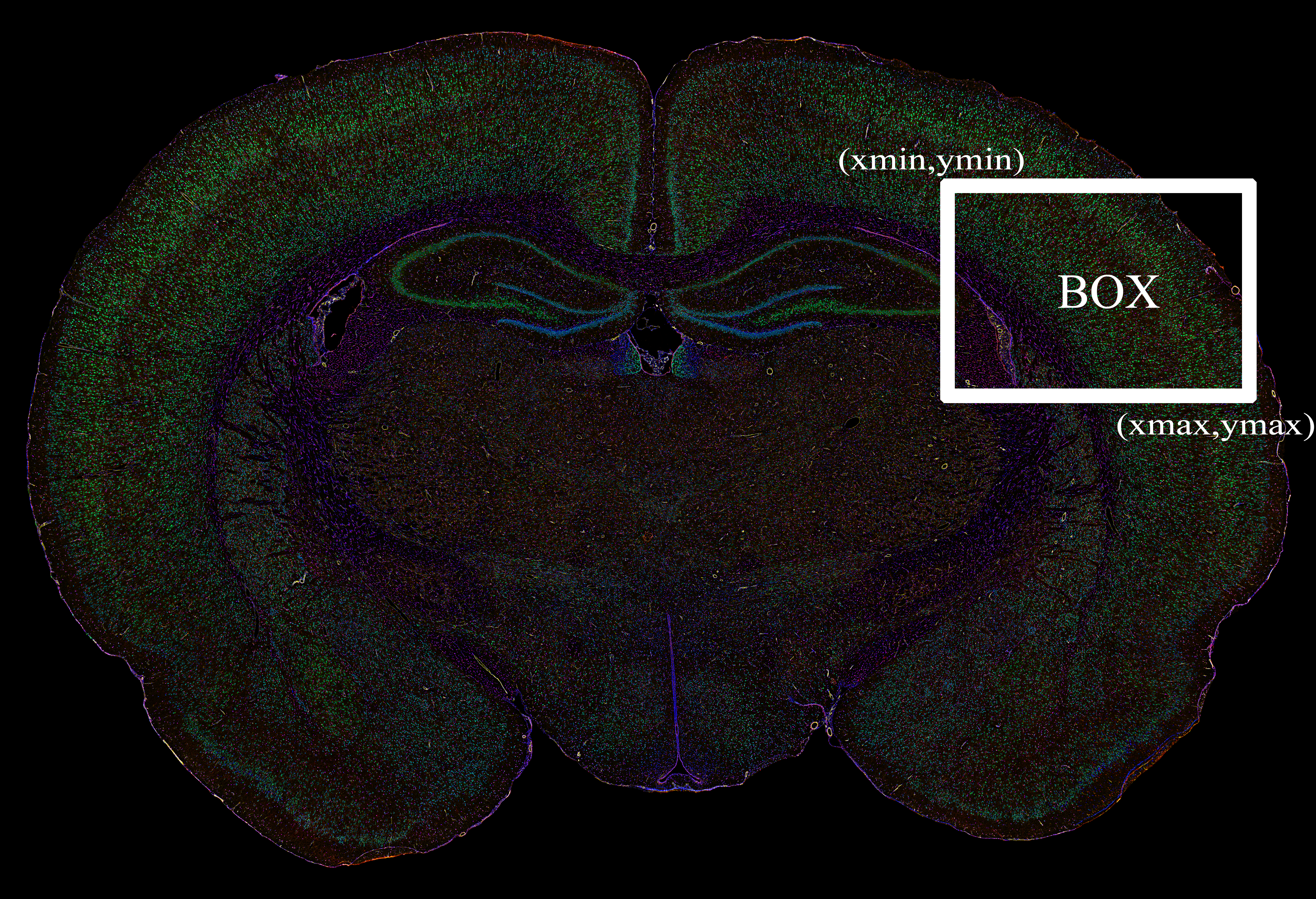
BRIGHTFIELD: Number of Brightfield channel orNone
Will generate a script in
OUTPUT_DIR/script.csvlike follows:filename biomarker intra channel correction inter channel correction channel 1 channel 2 channel 3 xmin ymin xmax ymax R1C1.tif Yes Yes 34000 8000 44000 15000 R1C10.tif Yes Yes R1C7.tif R1C5.tif 34000 8000 44000 15000 R1C11.tif Brightfield No No 34000 8000 44000 15000 R1C2.tif Yes Yes R1C1.tif 34000 8000 44000 15000 R1C3.tif Yes Yes 34000 8000 44000 15000 R1C4.tif Yes Yes R1C1.tif 34000 8000 44000 15000 R1C5.tif Yes Yes R1C6.tif 34000 8000 44000 15000 R1C6.tif Yes Yes R1C5.tif 34000 8000 44000 15000 R1C7.tif Yes Yes R1C10.tif R1C5.tif 34000 8000 44000 15000 R1C8.tif Yes Yes R1C7.tif 34000 8000 44000 15000 R1C9.tif Yes Yes 34000 8000 44000 15000 R2C1.tif Yes Yes 34000 8000 44000 15000 R2C10.tif Yes Yes R2C8.tif 34000 8000 44000 15000 R2C11.tif Brightfield No No 34000 8000 44000 15000 R2C2.tif Yes Yes 34000 8000 44000 15000 R2C3.tif Yes Yes R2C8.tif R2C6.tif 34000 8000 44000 15000 R2C4.tif Yes Yes 34000 8000 44000 15000 R2C5.tif Yes Yes R2C3.tif R2C6.tif 34000 8000 44000 15000 R2C6.tif Yes Yes R2C3.tif R2C8.tif 34000 8000 44000 15000 R2C7.tif Yes Yes R2C6.tif R2C8.tif R2C5.tif 34000 8000 44000 15000 R2C8.tif Yes Yes 34000 8000 44000 15000 R2C9.tif Yes Yes 34000 8000 44000 15000 -
if
supervised:SCRIPT: Path to the updatedscript.csvfile for reconstruction configuration:
filename biomarker intra channel correction inter channel correction channel 1 channel 2 channel 3 xmin ymin xmax ymax R1C1.tif DAPI Yes No 34000 8000 44000 15000 R1C10.tif NFH No Yes R1C7.tif 34000 8000 44000 15000 R1C11.tif Brightfield No No 34000 8000 44000 15000 R1C2.tif DAPI Yes No 34000 8000 44000 15000 R1C3.tif CC3 Yes No 34000 8000 44000 15000 R1C4.tif NeuN Yes No 34000 8000 44000 15000 R1C5.tif MBP No Yes R1C6.tif 34000 8000 44000 15000 R1C6.tif RECA1 Yes Yes R1C5.tif R1C8.tif 22000 24000 32000 31000 R1C7.tif IBA1 Yes Yes R1C10.tif R1C5.tif R1C8.tif 34000 8000 44000 15000 R1C8.tif TomatoLectin Yes Yes R1C7.tif 34000 8000 44000 15000 R1C9.tif PCNA Yes No 34000 8000 44000 15000 R2C1.tif DAPI Yes No 34000 8000 44000 15000 R2C10.tif MAP2 No Yes R2C8.tif 34000 8000 44000 15000 R2C11.tif Brightfield No No 34000 8000 44000 15000 R2C2.tif DAPI Yes No 34000 8000 44000 15000 R2C3.tif GAD67 Yes Yes R2C8.tif R2C6.tif 34000 16000 44000 23000 R2C4.tif GFAP Yes No 34000 8000 44000 15000 R2C5.tif Parvalbumin Yes Yes R1C5.tif R2C6.tif 31000 12000 41000 19000 R2C6.tif S100 Yes Yes R2C3.tif R2C8.tif 34000 8000 44000 15000 R2C7.tif Calretinin Yes Yes R2C6.tif R2C8.tif 18000 1000 28000 17000 R2C8.tif TomatoLectin Yes No 34000 8000 44000 15000 R2C9.tif CD31 Yes No 34000 8000 44000 15000

Parse the arguments to main_detection.py:
- if only DAPI:
python main_detection.py \ --INPUT_DIR /path/to/input/dir \ --OUTPUT_DIR /path/to/output/dir \ --DAPI R2C1.tif
- if DAPI + Histones:
python main_detection.py \ --INPUT_DIR /path/to/input/dir \ --OUTPUT_DIR /path/to/input/dir \ --DAPI R2C1.tif \ --HISTONES R2C2.tif
Parse the arguments to main_classification.py:
- if first time classifying:
python main_classification.py \ --INPUT_DIR /path/to/input/dir \ --OUTPUT_DIR /path/to/output/dir \ --BBXS_FILE /path/to/bbxs_detection.txt \ --DAPI R2C1.tif \ --HISTONES R2C2.tif \ --NEUN R2C4.tif \ --S100 R3C5.tif \ --OLIG2 R1C9.tif \ --IBA1 R1C5.tif \ --RECA1 R1C6.tif \ --test_mode first \ --thresholds 0.5 0.5 0.5 0.5 0.5
- if you want to adjust the classification results based on new thresholds:
--INPUT_DIR /path/to/input/dir \ --OUTPUT_DIR /path/to/output/dir \ --BBXS_FILE /path/to/bbxs_detection.txt \ --DAPI R2C1.tif \ --HISTONES R2C2.tif \ --NEUN R2C4.tif \ --S100 R3C5.tif \ --OLIG2 R1C9.tif \ --IBA1 R1C5.tif \ --RECA1 R1C6.tif \ --test_mode adjust \ --thresholds .5 .5 .5 .8 .5
You can update the classification table by using .fcs or .ice files in FCS Express or
Kaluza software to apply real time gating for phenotyping.
Using bounding boxes generated from detection module you can generate .fcs and .ice files.
Parse the arguments to GenerateICE_FCS_script.py:
- From bbox
python PHENOTYPING/GenerateICE_FCS_script.py \ --INPUT_DIR /path/to/input/dir \ --OUTPUT_DIR /path/to/output/dir \ --maskType=b \ --maskDir /path/to/bbxs_detection.txt \ --CHNDEF /path/to/channel/description/50_plex.csv \ --downscaleRate 4 \ --seedSize 2 \ --erosion_px 5
Arguments:
INPUT_DIR: Path to the directory containing input imagesOUTPUT_DIR: Path to the directory saving output imagesmaskType: Cell detection inputsmaskorbboxCHNDEF: dataset definition .csv filedownscaleRate: for FCSexpress like visulization software,downscale the image to avoid crashingseedSize: size of nuclear seed objectserosion_px: pixel to shrink the bbox to focus on nuclear
- Astrocyte Mask Generation:
To get soma, processes and whole cell masks for astrocytes using S100 and GFAP biomarkers run the following:
matlab -nodesktop -nosplash -r "astrocytes_whole_brain_segmentation('DAPI_PATH','E:\50-plex\final\S1_R1C1.tif', 'HISTONE_PATH','E:\50-plex\final\S1_R2C2.tif','S100_PATH','E:\50-plex\final\S1_R3C5.tif','GFAP_PATH', 'E:\50-plex\final\S1_R3C3.tif','OUTPUT_DIR','astrocytes_OUTPUT','CLASSIFICATION_table_path', 'E:\50-plex\classification_results\classification_table.csv','SEGMENTATION_MASKS','data/merged_labelmask.txt')"
Arguments:
-
DAPI_PATH: Path to DAPI channel -
HISTONE_PATH: Path to Histone channel -
GFAP_PATH: Path to GFAP channel -
S100_PATH: Path to S100 channel -
OUTPUT_DIR: Path to output masks -
CLASSIFICATION_table_path: Path to classification table as in 3 -
SEGMENTATION_MASKS: Path to segmentation masks -
Endothelials Mask Generation:
To get soma, processes and whole cell masks for endothelials using GFP and RECA1 biomarkers run the following:
matlab -nodesktop -nosplash -r "endothelial_whole_brain_segmentation('DAPI_PATH','E:\50-plex\final\S1_R1C1.tif','HISTONE_PATH','E:\50-plex\final\S1_R2C2.tif','GFP_PATH','E:\50-plex\final\S1_R1C4.tif','RECA1_PATH','E:\50-plex\final\S1_R1C6.tif','OUTPUT_DIR','endothelial_OUTPUT','CLASSIFICATION_table_path','E:\50-plex\classification_results\classification_table.csv','SEGMENTATION_MASKS','data/merged_labelmask.txt')"
Arguments:
-
DAPI_PATH: Path to DAPI channel -
HISTONE_PATH: Path to Histone channel -
GFP_PATH: Path to GFP channel -
RECA1_PATH: Path to RECA1 channel -
OUTPUT_DIR: Path to output masks -
CLASSIFICATION_table_path: Path to classification table as in 3 -
SEGMENTATION_MASKS: Path to segmentation masks -
Microglia Mask Generation:
To get soma, processes and whole cell masks for microglia using IBA1 biomarkers run the following:
matlab -nodesktop -nosplash -r "microglia_whole_brain_segmentation('DAPI_PATH','E:\50-plex\final\S1_R1C1.tif','HISTONE_PATH','E:\50-plex\final\S1_R2C2.tif','IBA1_PATH','E:\50-plex\final\S1_R1C5.tif','OUTPUT_DIR','microglia_OUTPUT','CLASSIFICATION_table_path','E:\50-plex\classification_results\classification_table.csv','SEGMENTATION_MASKS','data/merged_labelmask.txt')"
Arguments:
-
DAPI_PATH: Path to DAPI channel -
HISTONE_PATH: Path to Histone channel -
IBA1_PATH: Path to IBA1 channel -
OUTPUT_DIR: Path to output masks -
CLASSIFICATION_table_path: Path to classification table as in 3 -
SEGMENTATION_MASKS: Path to segmentation masks -
Neuronal Mask Generation:
To get soma, processes and whole cell masks for neurons using NeuN and MAP2 biomarkers run the following:
matlab -nodesktop -nosplash -r "neuron_whole_brain_segmentation('DAPI_PATH','E:\50-plex\final\S1_R1C1.tif','HISTONE_PATH','E:\50-plex\final\S1_R2C2.tif','NeuN_PATH','E:\50-plex\final\S1_R2C4.tif','MAP2_PATH','E:\50-plex\final\S1_R5C9.tif','OUTPUT_DIR','neuron_OUTPUT','CLASSIFICATION_table_path','E:\50-plex\classification_results\classification_table.csv','SEGMENTATION_MASKS','data/merged_labelmask.txt')"
Arguments:
-
DAPI_PATH: Path to DAPI channel -
HISTONE_PATH: Path to Histone channel -
NeuN_PATH: Path to NeuN channel -
MAP2_PATH: Path to MAP2 channel -
OUTPUT_DIR: Path to output masks -
CLASSIFICATION_table_path: Path to classification table as in 3 -
SEGMENTATION_MASKS: Path to segmentation masks -
Oligodendrocyte Mask Generation:
To get soma, processes and whole cell masks for oligodendrocytes using Olig2 and CNPase biomarkers run the following:
matlab -nodesktop -nosplash -r "oligodendrocytes_whole_brain_segmentation('DAPI_PATH','E:\50-plex\final\S1_R1C1.tif','HISTONE_PATH','E:\50-plex\final\S1_R2C2.tif','OLIG2_PATH','E:\50-plex\final\S1_R1C5.tif','CNPASE_PATH','E:\50-plex\final\S1_R5C4.tif','OUTPUT_DIR','oligodendrocytes_OUTPUT','CLASSIFICATION_table_path','E:\50-plex\classification_results\classification_table.csv','SEGMENTATION_MASKS','data/merged_labelmask.txt')"
Arguments:
DAPI_PATH: Path to DAPI channelHISTONE_PATH: Path to Histone channelOLIG2_PATH: Path to Olig2 channelCNPASE_PATH: Path to CNPase channelOUTPUT_DIR: Path to output masksCLASSIFICATION_table_path: Path to classification table as in 3SEGMENTATION_MASKS: Path to segmentation masks
The sample 50_plex dataset in a windows machine with the following configuration:
- 32 core Intel(R) Xeon(R) CPU E5-2687W 0 @ 3.10GHz
- NVIDIA GeForce GTX 1080 Ti
- 256 GB RAM
The run time for each module is:
- RECONSTRUCTION: 6 hours
- DETECTION: 4 hours
- CLASSIFICATION: 2 hours
- PHENOTYPING: 1 hour
- MORPHOLOGICAL MASKING: 4 hours
If you found this implementation useful in your work, please cite the paper:
@article{maric2021whole,
title={Whole-brain tissue mapping toolkit using large-scale highly multiplexed immunofluorescence imaging and deep neural networks},
author={Maric, Dragan and Jahanipour, Jahandar and Li, Xiaoyang Rebecca and Singh, Aditi and Mobiny, Aryan and Van Nguyen, Hien and Sedlock, Andrea and Grama, Kedar and Roysam, Badrinath},
journal={Nature communications},
volume={12},
number={1},
pages={1--12},
year={2021},
publisher={Nature Publishing Group}
}