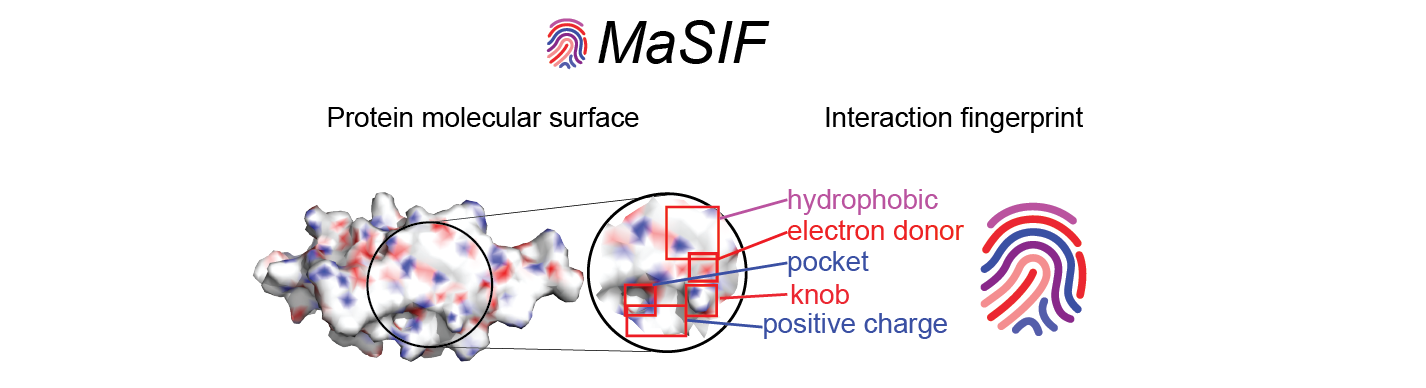
MaSIF- Molecular Surface Interaction Fingerprints: Geometric deep learning to decipher patterns in protein molecular surfaces.
- Description
- System and hardware requirements
- Software prerequisites
- Installation
- Method overview
- MaSIF applications
- PyMOL plugin
- License
- Reference
MaSIF is a proof-of-concept method to decipher patterns in protein surfaces important for specific biomolecular interactions. To achieve this, MaSIF exploits techniques from the field of geometric deep learning. First, MaSIF decomposes a surface into overlapping radial patches with a fixed geodesic radius, wherein each point is assigned an array of geometric and chemical features. MaSIF then computes a descriptor for each surface patch, a vector that encodes a description of the features present in the patch. Then, this descriptor can be processed in a set of additional layers where different interactions can be classified. The features encoded in each descriptor and the final output depend on the application-specific training data and the optimization objective, meaning that the same architecture can be repurposed for various tasks.
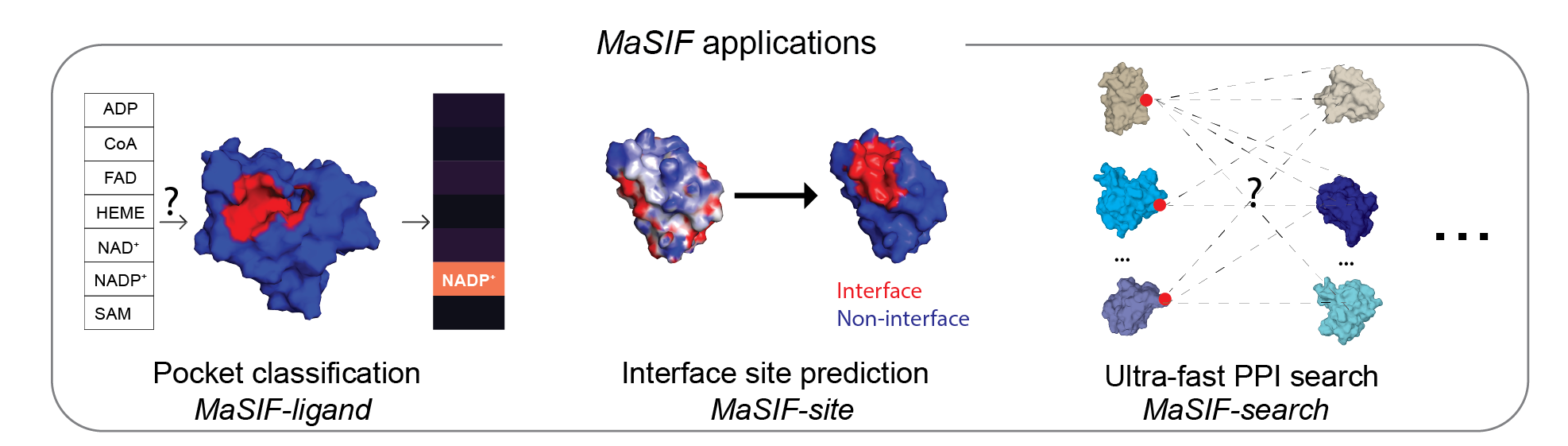
This repository contains a protocol to prepare protein structure files into feature-rich surfaces (with both geometric and chemical features), to decompose these into patches, and tensorflow-based neural network code to identify patterns in these using deep geometric learning. To show the potential of the approach, we showcase three proof-of-concept applications: a) ligand prediction for protein binding pockets (MaSIF-ligand); b) protein-protein interaction (PPI) site prediction in protein surfaces, to predict which surface patches on a protein are more likely to interact with other proteins (MaSIF-site); c) ultrafast scanning of surfaces, where we use surface fingerprints from binding partners to predict the structural configuration of protein-protein complexes (MaSIF-search).
This repository reproduces the experiments of:
Gainza, P., Sverrisson, F., Monti, F., Rodola, E., Bronstein, M. M., & Correia, B. E. (2019). Deciphering interaction fingerprints from protein molecular surfaces. bioRxiv, 606202.
MaSIF is distributed under an Apache License. This code is meant to serve as a tutorial, and the basis for researchers to exploit MaSIF in protein-surface learning tasks.
MaSIF has been tested on both Linux (Red Hat Enterprise Linux Server release 7.4, with a Intel(R) Xeon(R) CPU E5-2650 v2 @ 2.60GHz processesor and 16GB of memory allotment) and Mac OS environments (macOS High Sierra, processor 2.8 GHz Intel Core i7, 16GB memory). To reproduce the experiments in the paper, the entire datasets for all proteins consume about 1.4 terabytes.
Currently, MaSIF takes about 2 minutes to preprocess every protein. For this reason, we recommend a distributed cluster to preprocess the data for large datasets of proteins. Once data has been preprocessed, we strongly recommend using a GPU to train or evaluate the trained models as it can be up to 100 times faster than a CPU.
MaSIF relies on external software/libraries to handle protein databank files and surface files, to compute chemical/geometric features and coordinates, and to perform neural network calculations. The following is the list of required libraries and programs, as well as the version on which it was tested (in parenthesis).
- Python (2.7)
- reduce (3.23). To add protons to proteins.
- MSMS (2.6.1). To compute the surface of proteins.
- BioPython (1.66) . To parse PDB files.
- PyMesh (0.1.14). To handle ply surface files, attributes, and to regularize meshes.
- pyflann (1.6.14). To perform nearest neighbor searches of vertices.
- PDB2PQR (2.1.1), multivalue, and APBS (1.5). These programs are necessary to compute electrostatics charges.
- open3D (0.5.0.0). Mainly used for RANSAC alignment.
- matlab (R2018a). Used to compute some geometric features and angular/radial coordinates.
- Python bindings for matlab - To call matlab functions from within Python.
- Tensorflow (1.9). Use to model, train, and evaluate the actual neural networks. Models were trained and evaluated on a NVIDIA Tesla K40 GPU.
- SBI. Used for parsing PDB files and generate biological assembly for MaSIF-ligand.
- Dask (2.2.0). Run function calls on multiple threads (Optional for reproducing some benchmarks).
- Pymol. This optional plugin allows one to visualize surface files in PyMOL.
We are working to reduce this list of requirements for future versions.
After preinstalling dependencies, add the following environment variables to your path, changing the appropriate directories:
export APBS_BIN=/path/to/apbs/APBS-1.5-linux64/bin/apbs
export MULTIVALUE_BIN=/path/to/apbs/APBS-1.5-linux64/share/apbs/tools/bin/multivalue
export PDB2PQR_BIN=/path/to/apbs/apbs/pdb2pqr-linux-bin64-2.1.1/pdb2pqr
export PATH=$PATH:/path/to/reduce/
export REDUCE_HET_DICT=/path/to/reduce/reduce_wwPDB_het_dict.txt
export PYMESH_PATH=/path/to/PyMesh
export MSMS_BIN=/path/to/msms/msms
export PDB2XYZRN=/path/to/msms/pdb_to_xyzrn
Clone masif to a local directory
git clone https://github.com/lpdi-epfl/masif
cd masif/
Since MaSIF is written in Python and Matlab, no compilation is required.
From a protein structure MaSIF computes a molecular surface discretized as a mesh according to the solvent excluded surface (computed using MSMS), and assigns geometric and chemical features to every point (vertex) in the mesh. Around each vertex of the mesh, we extract a patch with geodesic radius of r=9 Å or r=12 Å. Then, MaSIF applies a geometric deep neural network to these patches. The neural network consists of one or more layers applied sequentially; a key component of the architecture is the geodesic convolution, generalizing the classical convolution to surfaces and implemented as an operation on local patches.
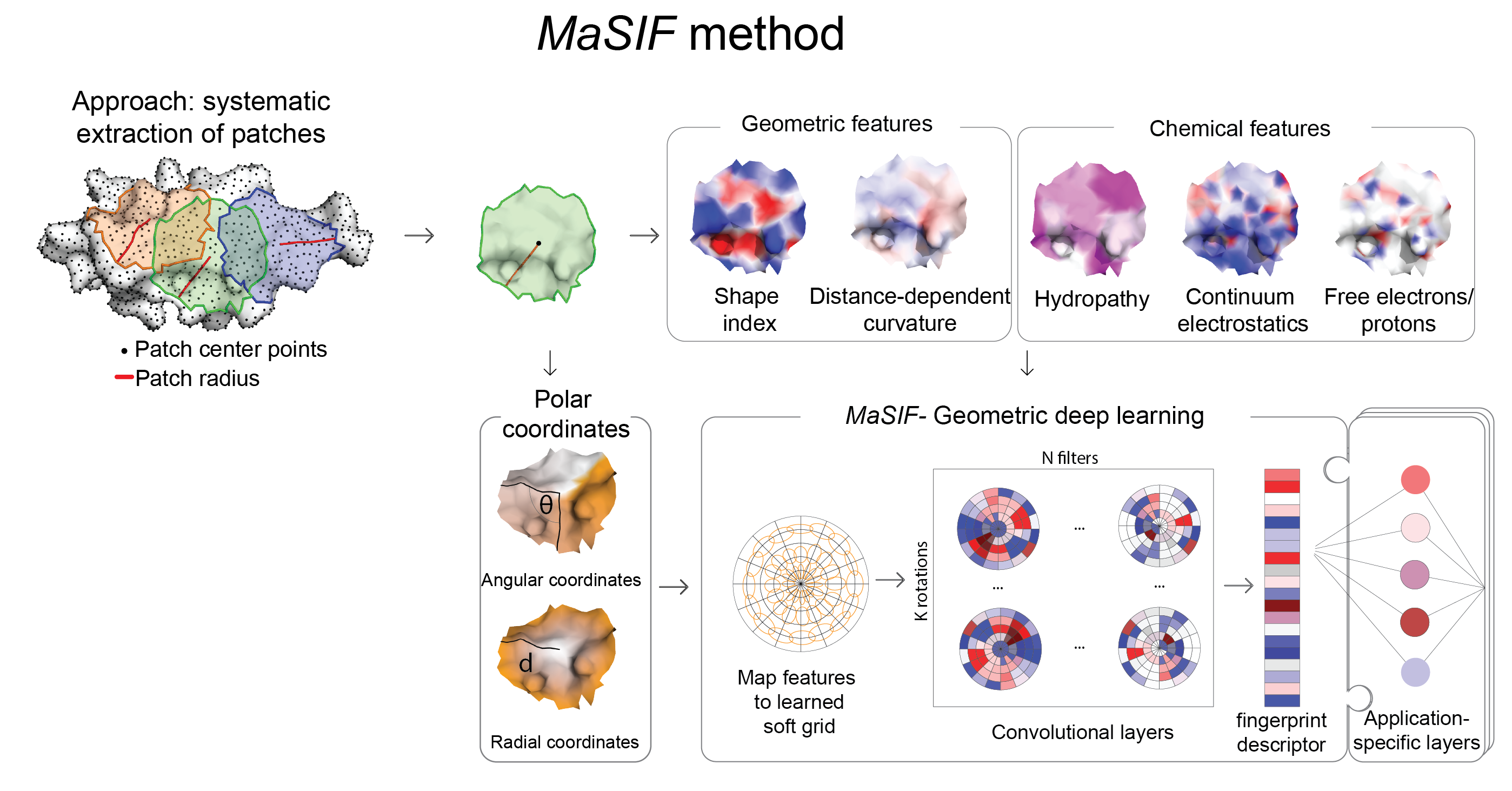
The procedure is repeated for different patch locations similarly to a sliding window operation on images, producing the surface fingerprint descriptor at each point, in the form of a vector that stores information about the surface patterns of the center point and its neighborhood. The parameter set minimizes a cost function on the training dataset, which is specific to each application that we present here.
For each application, MaSIF requires a preprocessing of data. This entails a running a scripted protocol, which performs the following steps:
- Download the PDB.
- Protonate the PDB, extract the desired chains, triangulate the surface (using MSMS), and compute chemical features.
- Translate the surface to a matlab file and compute the shape index on each vertex.
- Compute the angular and radial geodesic coordinates for each patch.
- Extract all patches, with features and coordinates, for each protein.
Each application data directory (under masif/data/masif*) contains a script to precompute the data.
To run this protocol for a single protein, (e.g. chain A of PDB id code 1MBN ) run:
./data_prepare_one.sh 1MBN_A_
To run it on a pair of interacting protein domains (chains A,B, of PDB id 1AKJ form the first domain and chains D,E form the second domain):
./data_prepare_one.sh 1AKJ_AB_DE
If you have access to a cluster (strongly recommended), then this process can be run in parallel. If your cluster supports slurm files, we provide a slurm file under each application data directory. which can be run using sbatch:
sbatch data_prepare.slurm
Most of the PDBs that were used for the paper, and their corresponding surfaces (with precomputed chemical features) are available at: https://doi.org/10.5281/zenodo.2625420 . The unbound proteins are available in this repository under data/masif_ppi_search_ub/data_preparation/00-raw_pdbs/.
Note that the preparation of the data can consume a large amount of space for large protein databases. This is due to the fact that the preprocessing step decomposes protein surfaces into overlapping patches, which results in a large amount of duplicated data. In upcoming versions we hope to optimize this process to perform patch-decomposition operations on-the-fly, eliminating the need for large amounts of disk space.
MaSIF was tested on three proof-of-concept applications. For each application we provide the trained neural network model that was used for the main experiments in the paper.
cd data/masif_site/
The lists of pdb ids and chains used in the training and test sets are located, in numpy format, under:
data/masif_ligand/lists/test_pdbs_sequence.npy
data/masif_ligand/lists/train_pdbs_sequence.npy
data/masif_ligand/lists/val_pdbs_sequence.npy
Each of these files can be read using the numpy.load function.
Precompute the datasets (see MaSIF data preparation), ideally using slurm:
sbatch prepare_data.slurm
Be sure you have enough disk space, about 400GB.
Once the data has been precomputed, MaSIF-ligand requires the generation of Tensorflow TFRecords for training. For this, either run slurm or execute the command present in the make_tfrecord.slurm file:
sbatch make_tfrecord.slurm
Once the tfrecords have been precomputed, the training for the network can start, where we strongly recommend a GPU (run the commands in the slurm file one by one if you do not have slurm):
sbatch train_model.slurm
To evaluate the neural network run:
sbatch evaluate_test.slurm
The output of the evaluation is placed under the data/masif_ligand/test_set_predictions/ directory, with two numpy files per input protein databank structure, e.g.:
5LXM_AD_labels.npy
5LXM_AD_logits.npy
where the labels file contains the ground truth, and the logits file contains the prediction logits.
Change to the masif-site data directory.
cd data/masif_site/
The lists of pdb ids and chains used in the training and test sets are located under:
data/masif_site/data/lists/full_list.txt
data/masif_site/data/lists/training.txt
data/masif_site/data/lists/testing.txt
Precompute the datasets (see MaSIF data preparation), ideally using slurm:
sbatch prepare_data.slurm
Be sure you have enough disk space, about 400GB.
Once the data has been precomputed, the training for the network can start:
./train_nn.sh
For the experiments in the paper we trained MaSIF-site for 40 hours.
Once a network has been trained, specific proteins can be evaluated. For example to evaluate the selected subset of transient interactions:
./predict_site.sh
The predictions for each vertex in each protein are stored in the directory data/masif_site/output/all_feat_3l/pred_data/. The surfaces of the predicted sites can be colored according to the site prediction:
./color_site.sh
and saved to a ply file, under the directory: data/masif_site/output/all_feat_3l/pred_surfaces/
These surfaces can then be visualized using our PyMOL plugin.
A jupyter notebook with code to compare the prediction on the transient interactiosn of this test set to the program SPPIDER can be found at:
masif/comparison/masif_site/masif_vs_sppider/masif_sppider_comp.ipynb
Change to the masif-search data directory.
cd data/masif_ppi_search/
The lists of pdb ids and chains used in the training and test sets are located under:
data/masif_ppi_searhc/data/lists/full_list.txt
data/masif_site/data/lists/training.txt
data/masif_site/data/lists/testing.txt
Precompute the datasets (see MaSIF data preparation), ideally using slurm:
sbatch prepare_data.slurm
Be sure you have enough disk space, about 400GB.
For speed reasons, the actual data that will be used by the neural network is cached in a separate directory. This data consists of the pairs of patches that pass a shape complementarity threshold and an equal number of random patches. This process is run by executing:
./cache_nn.sh nn_models.sc05.custom_params
Once the data has been cached, the training for the network can start:
./train.sh nn_models.sc05.custom_params
For the paper we trained for about 40 hours. The neural network model is saved in the nn_models/sc05/all_feat/model_data directory whenever the validation ROC AUC improves over the previously saved model's validation ROC AUC.
Once the neural network has been trained and saved, descriptors for specific proteins can be computed using the command:
./compute_descriptors.sh lists/testing.txt
These descriptors are saved under the descriptors/ directory.
To evaluate the second stage ransac protocol, go to the masif/comparison/masif_ppi_search directory:
cd $masif_root/comparison/masif_ppi_search/masif_descriptors/
./second_stage.sh
To reproduce the large PD-L1:PD1 benchmark presented in the paper:
cd data/masif_ppi_search/pdl1_benchmark
./run_benchmark.sh
A PyMOL plugin to visualize protein surfaces is provided in the source/pymol subdirectory. We used this plugin for all the structural figures shown in our paper. This plugin requires PyMOL and PyMesh to be installed in your local computer.
To install the plugin go to the Plugin -> Plugin Manager window in PyMOL and choose the Install new plugin tab. Then select the masif/source/masif_pymol_plugin.zip file.
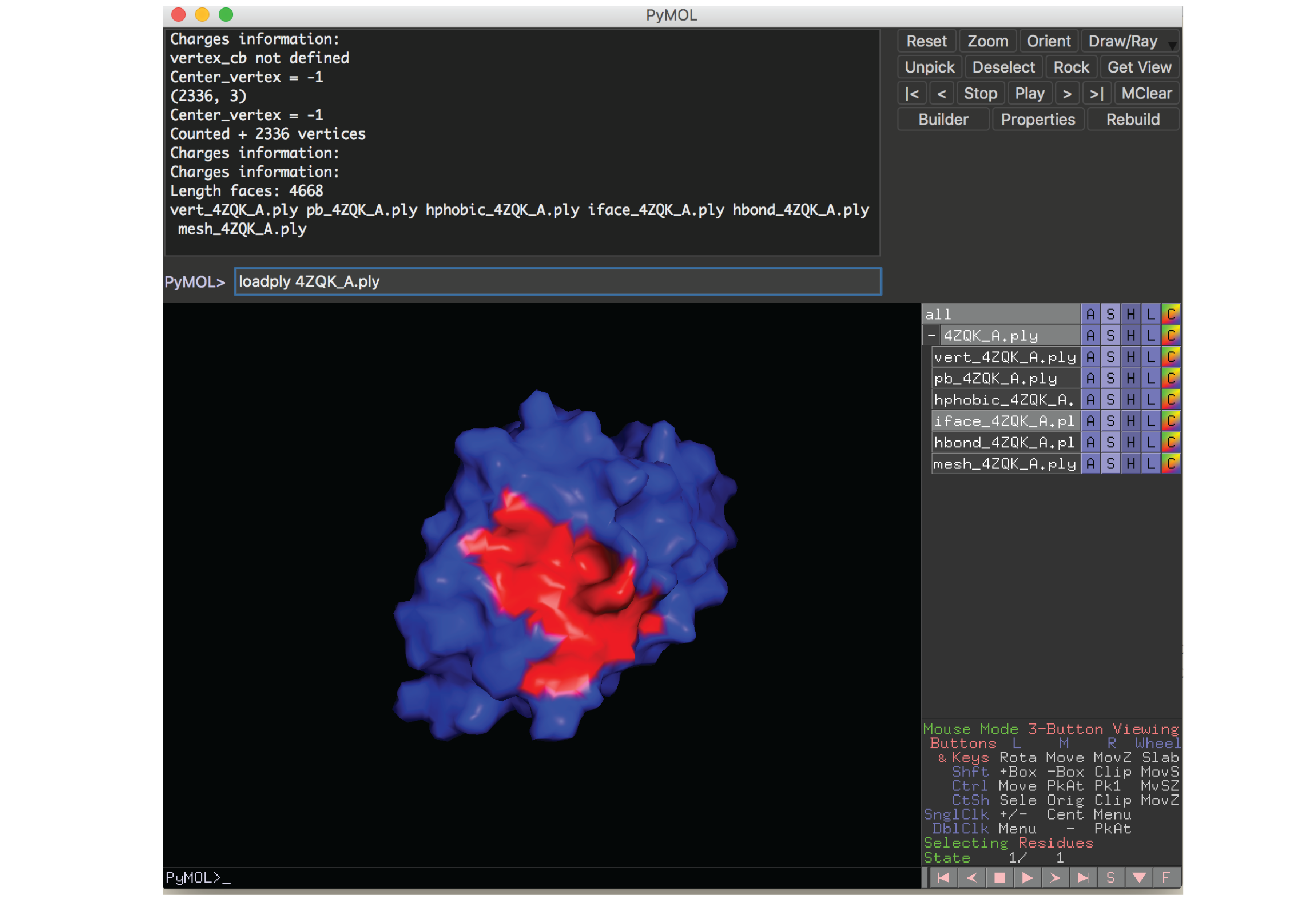
To load a protein surface file, run this command inside PyMOL:
loadply ABCD_E.ply
Example:
MaSIF is released under an Apache v2.0 license.
If you use this code, please use the bibtex entry in citation.bib