![]()
Caretta is a software-suite to perform multiple protein structure alignment and structure feature extraction.
The older, slower version of Caretta as described in https://doi.org/10.1016/j.csbj.2020.03.011 can be found at https://git.wur.nl/durai001/caretta
- Linux and Mac
- All capabilities are supported
- Windows
- The external tool msms is not available in Windows. Due to this:
- Feature extraction is not available.
featuresargument in caretta-cli must always be run with--only-dssp.caretta-appis not available.
Caretta works with Python 3.9+ Run the following commands to install external dependencies required for feature extraction (Mac and Linux only) This is not required if you are only using the alignment.
conda install -c salilab dssp
conda install -c bioconda msms
pip install git+https://github.com/TurtleTools/caretta.gitpip install "caretta[GUI] @ git+https://github.com/TurtleTools/caretta.git"export OMP_NUM_THREADS=1 # this should always be 1
export NUMBA_NUM_THREADS=20 # change to required number of threadscaretta-cli input_pdb_folder
# e.g. caretta-cli test_data Options:
Usage: caretta-cli [OPTIONS] INPUT_PDB [MODEL_FOLDER]
Align protein structures using Caretta.
Writes the resulting sequence alignment and superposed PDB files to
"caretta_results". Optionally also outputs a set of aligned feature
matrices, or the python class with intermediate structures made during
progressive alignment.
Arguments:
INPUT_PDB A folder with input protein files [required]
Options:
-p FLOAT gap open penalty [default: 1.0]
-e FLOAT gap extend penalty [default: 0.01]
-c, --consensus-weight weight well-aligned segments to reduce gaps
in these areas [default: True]
-f, --fast Don't use all vs. all pairwise alignment for
distance matrix calculation
-o, --output PATH folder to store output files [default:
caretta_results]
--fasta / --no-fasta write alignment in FASTA file format
[default: fasta]
--pdb / --no-pdb write PDB files superposed according to
alignment [default: pdb]
-t, --threads INTEGER number of threads to use [default: 4]
--features extract and write aligned features as a
dictionary of NumPy arrays into a pickle
file
--only-dssp extract only DSSP features
--class write StructureMultiple class with
intermediate structures and tree to pickle
file
--matrix write pre-aligned distance matrix and post-
aligned RMSD, coverage and TM-score matrices
to files
-v, --verbose Control verbosity [default: True]
--install-completion [bash|zsh|fish|powershell|pwsh]
Install completion for the specified shell.
--show-completion [bash|zsh|fish|powershell|pwsh]
Show completion for the specified shell, to
copy it or customize the installation.
--help Show this message and exit.
caretta-app <host-ip> <port>
# e.g. caretta-app localhost 8091Then go to localhost:8091/caretta in a browser window.
Read features as:
import pickle
with open('caretta_results/result_features.pkl', 'rb') as f:
protein_ids, features = pickle.load(f)The features dictionary is a dictionary of NumPy arrays, with the protein_ids defining the order of rows, the
columns being the aligned feature values, and the keys as follows:
dssp_NH_O_1_index,dssp_NH_O_1_energy,dssp_NH_O_2_index,dssp_NH_O_2_energy,dssp_O_NH_1_index,dssp_O_NH_1_energy,dssp_O_NH_2_index,dssp_O_NH_2_energy: hydrogen bonds; e.g. -3,-1.4 means: if this residue is residue i then N-H of I is h-bonded to C=O of I-3 with an electrostatic H-bond energy of -1.4 kcal/mol. There are two columns for each type of H-bond, to allow for bifurcated H-bonds.dssp_acc: number of water molecules in contact with this residue *10. or residue water exposed surface in Angstrom^2.dssp_alpha: virtual torsion angle (dihedral angle) defined by the four Cα atoms of residues I-1,I,I+1,I+2. Used to define chirality.dssp_kappa: virtual bond angle (bend angle) defined by the three Cα atoms of residues I-2,I,I+2. Used to define bend (structure code ‘S’).dssp_phi: IUPAC peptide backbone torsion angles.dssp_psi: IUPAC peptide backbone torsion angles.dssp_tco: cosine of angle between C=O of residue I and C=O of residue I-1. For α-helices, TCO is near +1, for β-sheets TCO is near -1.anm_ca: Fluctuations of alpha carbon atoms based on an Anisotropic network modelanm_cb: Fluctuations of beta carbon atoms based on an Anisotropic network modelgnm_ca: Fluctuations of alpha carbon atoms based on a Gaussian network modelgnm_cb: Fluctuations of beta carbon atoms based on a Gaussian network modeldepth_ca: Depths of alpha carbon atomsdepth_cb: Depths of beta carbon atomsdepth_mean: Mean depth of residues
Janani Durairaj, Mehmet Akdel, Dick de Ridder, Aalt DJ can Dijk. "Fast and adaptive protein structure representations for machine learning." Machine Learning for Structural Biology Workshop, NeurIPS 2020 (https://doi.org/10.1101/2021.04.07.438777)
Poster:
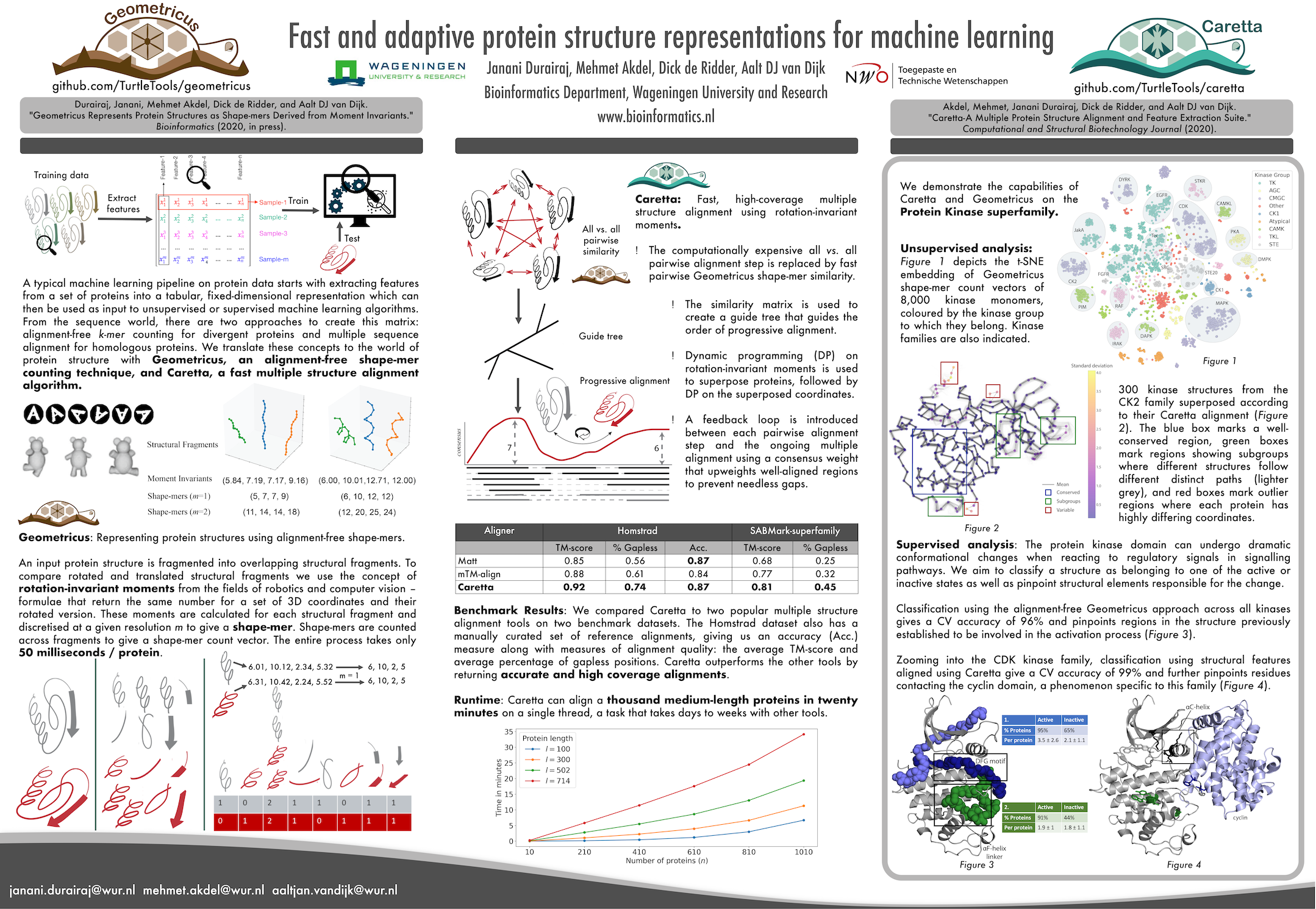
Akdel, Mehmet, Janani Durairaj, Dick de Ridder, and Aalt DJ van Dijk. "Caretta-A Multiple Protein Structure Alignment and Feature Extraction Suite." Computational and Structural Biotechnology Journal (2020) . (https://doi.org/10.1016/j.csbj.2020.03.011)