SEM is a nucleosome calling package, which focuses on characterizing nucleosome types genome-wide. The following nucleosome metrics would be produced in the output:
- Dyad location
- Occupancy
- Fuzziness
- Nucleosome subtype mixture probability
SEM distinguishes nucleosome type according to the DNA fragment length protected by the nucleosome. For example, a canonical nucleosome protects ~147bp DNA under extensive MNase digestion, while a hexamer should protect relatively shorter DNA. SEM assumes each type of nucleosome has its own distinct fragment length distribution, it deconvolves the fragment length profile of all DNA fragments to infer each distribution's parameters.
SEM is available on bioconda. To install, simply run:
$ conda install -c bioconda semAlso you can download the latest runnable JAR file from SEM releases.
Run SEM on a single MNase-seq experiment:
# clone the repo and run in the root directory
$ sem --expt data/test.bam --geninfo data/mm10.fa.fai --out test/ --numClusters 3 --threads 1The outputs will be in the test directory in this example.
Required:
--out <output file prefix>
--geninfo <genome info file>
--expt <file name> AND --format <SAM/BED/SCIDX>
OR
--design <experiment design file name to use instead of --expt and --ctrl>
General:
--threads <number of threads to use (default=1)>
--verbose [flag to print intermediate files and extra output]
--config <config file: all options here can be specified in a name<space>value text file, over-ridden by command-line args>
Genome:
--providedPotenialRegions <bed file to restrict nucleosome detection regions>
Loading Data:
--nonunique [flag to use non-unique reads]
--mappability <fraction of the genome that is mappable for these experiments (default=0.8)>
--nocache [flag to turn off caching of the entire set of experiments (i.e. run slower with less memory)]
Detecting nucleosome type:
--numClusters <number of nucleosome types>
--providedBindingSubtypes <custom binding subtypes (format: mean variance weight, sum of weights = 1)>
--onlyGMM <only Run GMM without the following nucleosome calling steps, use it to optimize nucleosome subtype parameters>
Running SEM:
--r <max. model update rounds, default=3>
--alphascale <alpha scaling factor(default=1.0>
--fixedalpha <minimum number of fragments a nucleosome should have (default=1, must >= 1)>
--exclude <file of regions to ignore, usually blacklist regions>
--consensusExclusion <consensus exclusion zone>
In SEM, Gaussian Mixture Model (GMM) is used on MNase-seq fragment size distribution to find the potential nucleosome subtypes, each nucleosome subtype is represented by a Normal Distribution with parameters mean and variance. When --numClusters is set as a positive integer, a finite GMM will be used to find out the parameters of each nucleosome subtype.
It's recommended to use Picard CollectInsertSizeMetrics first to check the distribution of fragment size distribution to decide the number of clusters. When there is no prior knowledge on the number of nucleosome subtypes, --numClusters can also be set as -1 to let SEM decide it by a Dirichlet Process Mixture Model (DPMM).
You can use --onlyGMM option to let SEM only run the nucleosome subtype characterization, the distribution of each nucleosome subtype would be plotted in intermediate-results directory in the results folder.
Users can also provide their own nucleosome subtype information instead of using SEM built-in functions by providing a file through --providedBindingSubtypes, file should be in the below format with tab delimited:
mean_1 var_1 weight_1
mean_2 var_2 weight_2
...
The sum of weights should be equal to 1
--r controls how many rounds of EM will be used, the default 3 rounds is able to return precise enough nucleosome predictions. This number can be increased to get more precise predictions but at the risk of overfitting.
--fixedalpha decides the threshold for nucleosome occupancy, during EM, all nucleosomes below this threshold will be terminated, which ensures all the remaining nucleosomes have occupancy >= fixedalpha
--consensusExclusion decides the exclusion zone between nucleosomes, the spacing between nucleosomes will be >= this threshold, deafult exclusion zone is 127bp.
Since nucleosomes are everywhere on the genome, it's both computational intensive and time consuming to do EM on all the nucleosomes, besides, not all nucleosomes are of interest sometimes. --providedPotenialRegions can accept a bed file to restrict the regions where SEM will do nucleosome finding in, an example of potential regions could be candidate cis-regulatory regions (ccREs) from ENCODE SCREEN project.
A potential issue when running SEM in this way is that large number of small pieces of regions could lead to imprecise nucleosome finding at the edge of regions. So it's recommended to expand each region then merge the regions close to each other, below is the comparison between original ccRE regions and merged ccRE regions by bedtools slop -b 500 and bedtools merge -d 500
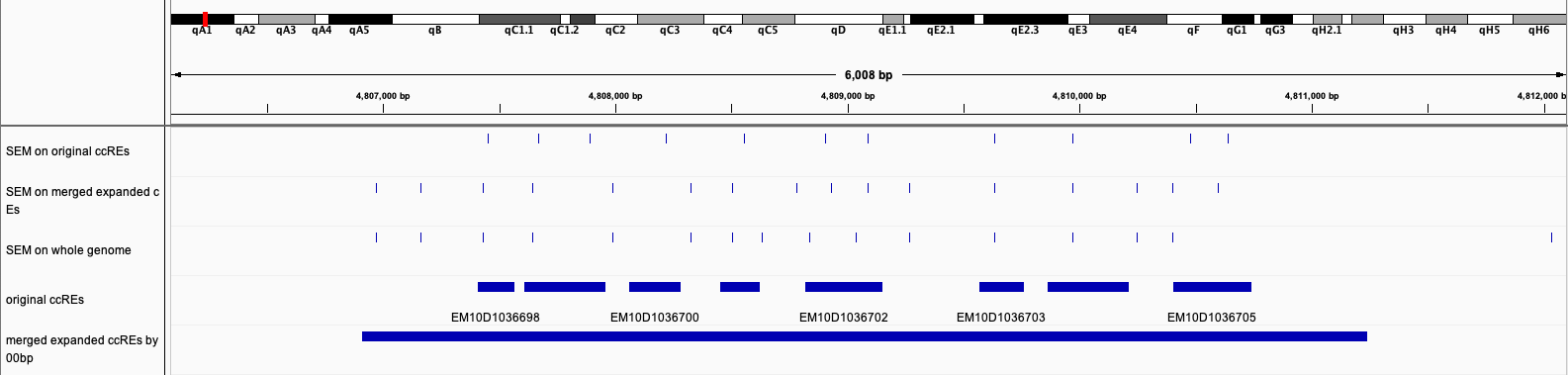
Note: even provided with a potential region file, SEM will still use all MNase-seq fragments on the genome to decide nucleosome subtypes, if you want a different behavior, please refer to Detecting nucleosome subtypes for how to provide customized nucleosome subtypes information.
Feel free to open new issues or contact the author Jianyu Yang (jmy5455@psu.edu) on usage or feature requests!