
![]()
aMeta is a Snakemake workflow for identifying microbial sequences in ancient DNA shotgun metagenomics samples. The workflow performs:
- trimming adapter sequences and removing reads shorter than 30 bp with Cutadapt
- quaity control before and after trimming with FastQC and MultiQC
- taxonomic sequence kmer-based classification with KrakenUniq
- sequence alignment with Bowtie2 and screening for common microbial pathogens
- deamination pattern analysis with MapDamage2
- Lowest Common Ancestor (LCA) sequence alignment with Malt
- authentication and validation of identified microbial species with MaltExtract
When using aMeta and / or pre-built databases provided together with the wokflow for your research projects, please cite our preprint: https://www.biorxiv.org/content/10.1101/2022.10.03.510579v1
- Nikolay Oskolkov (@LeandroRitter) nikolay.oskolkov@scilifelab.se
- Claudio Mirabello (@clami66) claudio.mirabello@scilifelab.se
- Per Unneberg (@percyfal) per.unneberg@scilifelab.se
Clone the github repository, then create and activate aMeta conda environment (here and below cd aMeta implies navigating to the cloned root aMeta directory). For this purpose, we recommend installing own conda, for example from here https://docs.conda.io/en/latest/miniconda.html, and mamba https://mamba.readthedocs.io/en/latest/installation.html:
git clone https://github.com/NBISweden/aMeta
cd aMeta
mamba env create -f workflow/envs/environment.yaml
# alternatively: conda env create -f workflow/envs/environment.yaml, however it takes longer
conda activate aMeta
Run a test to make sure that the workflow was installed correctly:
cd .test
./runtest.sh -j 4
Here, and below, by -j you can specify the number of threads that the workflow can use. Please make sure that the installation and test run accomplished successfully before proceeding with running aMeta on your data. Potential problems with installation and test run often come from unstable internet connection and particular conda settings used e.g. at computer clusters, therefore we advise you to use your own freshly installed conda.
To run the worflow you need to prepare a tab-delimited sample-file config/samples.tsv with at least two columns, and a configuration file config/config.yaml, below we provide examples for both files.
Here is an example of samples.tsv, this implies that the fastq-files are located in aMeta/data folder:
sample fastq
foo data/foo.fq.gz
bar data/bar.fq.gz
Currently, it is important that the sample names in the first column exactly match the names of the fastq-files in the second column. For example, a fastq-file "data/foo.fq.gz" specified in the "fastq" column, must have a name "foo" in the "sample" column. Please make sure that the names in the first and second columns match.
Below is an example of config.yaml, here you will need to download a few databases that we made public (or build databases yourself).
samplesheet: "config/samples.tsv"
# KrakenUniq Microbial NCBI NT database (if you are interested in prokaryotes only)
# can be downloaded from https://doi.org/10.17044/scilifelab.20518251
krakenuniq_db: resources/DBDIR_KrakenUniq_MicrobialNT
# KrakenUniq full NCBI NT database (if you are interested in prokaryotes and eukaryotes)
# can be downloaded from https://doi.org/10.17044/scilifelab.20205504
#krakenuniq_db: resources/DBDIR_KrakenUniq_Full_NT
# Bowtie2 index and helping files for following up microbial pathogens
# can be downloaded from https://doi.org/10.17044/scilifelab.21185887
bowtie2_db: resources/library.pathogen.fna
bowtie2_seqid2taxid_db: resources/seqid2taxid.pathogen.map
pathogenomesFound: resources/pathogensFound.very_inclusive.tab
# Bowtie2 index for full NCBI NT (for quick followup of prokaryotes and eukaryotes),
# can be downloaded from https://doi.org/10.17044/scilifelab.21070063 (please unzip files)
# For using Bowtie2 NT index, replace "bowtie2_db" and "bowtie2_seqid2taxid_db" above by
#bowtie2_db: resources/library.fna
#bowtie2_seqid2taxid_db: resources/seqid2taxid.map.orig
# Helping files for building Malt database
# can be downloaded from https://doi.org/10.17044/scilifelab.21070063
malt_nt_fasta: resources/library.fna
malt_seqid2taxid_db: resources/seqid2taxid.map.orig
malt_accession2taxid: resources/nucl_gb.accession2taxid
# A path for downloading NCBI taxonomy files (performed automatically)
# you do not need to change this line
ncbi_db: resources/ncbi
# Breadth and depth of coverage filters
# default thresholds are very conservative, can be tuned by users
n_unique_kmers: 1000
n_tax_reads: 200
There are several ways to download the database files. One option is to follow this link https://docs.figshare.com/#articles_search and search for the last number in the database links provided above in the "article_id" search bar. This will give you the download url for each file. Then you can either use wget inside a screen session or tmux session to download it, or aria2c, for example, https://aria2.github.io/. N.B. We strongly recommend you not to mix the databases in the same directory but place them in individual folders, otherwise they may overwrite each other. Also, if you use the KrakenUniq full NCBI NT database and / or Bowtie2 index of full NCBI NT, please keep in mind, that the reference genomes used for building the database / index were imported as is from the BLASTN tool https://blast.ncbi.nlm.nih.gov/Blast.cgi. This implies that the majority of eukaryotic reference genomes (including human reference genome) included in the database / index may be of poor quality for the sake of minimization of resource usage. In contrast, the vast majority of microbial reference genomes included in the NCBI NT database / index are of very good (complete) quality. Therefore, if the goal of your analysis is human / animal microbiome profiling, we recommend you to use the Microbial NCBI NT database / index, this will make sure that human / animal reads will not be acidentally assiged to microbial organisms. However, the full NCBI NT database / index are very useful if you work with e.g. sedimentary or environmental ancient DNA, and your goal is to simply detect in unbiased way all prokaryotic and eukaryotic organisms present in your samples, without trying to precisely quantify their abundance.
After you have prepared the sample- and configration-file, please install job-specific environments and update Krona taxonomy:
cd aMeta
snakemake --snakefile workflow/Snakefile --use-conda --conda-create-envs-only -j 20
env=$(grep krona .snakemake/conda/*yaml | awk '{print $1}' | sed -e "s/.yaml://g" | head -1)
cd $env/opt/krona/
./updateTaxonomy.sh taxonomy
cd -
Finally, the workflow can be run using the following command line:
cd aMeta
snakemake --snakefile workflow/Snakefile --use-conda -j 20
In the sections More configuration options, Environment module configuration and Runtime configuration we will give more information about fine-tuning the configuration as well as instructions on how to run the workflow in a computer cluster enviroment.
More details about running aMeta can be found in the step-by-step tutorial available in the aMeta/vignettes directory.
All output files of the workflow are located in aMeta/results directory. To get a quick overview of ancient microbes present in your samples you should check a heatmap in results/overview_heatmap_scores.pdf.

The heatmap demonstrates microbial species (in rows) authenticated for each sample (in columns). The colors and the numbers in the heatmap represent authentications scores, i.e. numeric quantification of eight quality metrics that provide information about microbial presence and ancient status. The authentication scores can vary from 0 to 10, the higher is the score the more likely that a microbe is present in a sample and is ancient. Typically, scores from 8 to 10 (red color in the heatmap) provide good confidence of ancient microbial presence in a sample. Scores from 5 to 7 (yellow and orange colors in the heatmap) can imply that either: a) a microbe is present but not ancient, i.e. modern contaminant, or b) a microbe is ancient (the reads are damaged) but was perhaps aligned to a wrong reference, i.e. it is not the microbe you think about. The former is a more common case scenario. The latter often happens when an ancient microbe is correctly detected on a genus level but we are not confident about the exact species, and might be aligning the damaged reads to a non-optimal reference which leads to a lot of mismatches or poor evennes of coverage. Scores from 0 to 4 (blue color in the heatmap) typically mean that we have very little statistical evedence (very few reads) to claim presence of a microbe in a sample.
To visually examine the eight quality metrics
- deamination profile,
- evenness of coverage,
- edit distance (amount of mismatches) for all reads,
- edit distance (amount of mismatches) for damaged reads,
- read length distribution,
- PMD scores distribution,
- number of assigned reads (depth of coverage),
- average nucleotide identity (ANI)
corresponding to the numbers and colors of the heatmap, one can find them in results/AUTHENTICATION/sampleID/taxID/authentic_Sample_sampleID.trimmed.rma6_TaxID_taxID.pdf for each sample sampleID and each authenticated microbe taxID. An example of such quality metrics is shown below:

In case you are interested in an overview of microbial species present in your samples irrespective of their ancient status, you can simply check a KrakenUniq abundance matrix here results/KRAKENUNIQ_ABUNDANCE_MATRIX/krakenuniq_absolute_abundance_heatmap.pdf:

The values in the heatmap above indicate the numbers of reads assigned to each microbe in each species. The corresponding Total Sum Scaled (TSS), aka library size normalized, KrakenUniq abundance matrix is located in results/KRAKENUNIQ_ABUNDANCE_MATRIX/krakenuniq_normalized_abundance_heatmap.pdf. Please note that the microbial species in the KrakenUniq abundance matrix might not always overlap with the ones present in the authentication score heatmap above. This is because not all microbes detected by KrakenUniq at the pre-screening step can be successfully validated by Malt + MaltExtract. The absolute and normalized microbial abundance heatmaps are optimal for visual exploration for up to ~50-100 samples and ~100-200 species. For larger numbers of samples and / or species, the heatmaps may become too crowded and difficult to view; therefore, in this case, the users are advised to utilize the raw KrakenUniq abundance matrix krakenuniq_abundance_matrix.txt for their own custom visualization.
Finally, below we list locations and provide short comments for a few other useful metrics / plots / information delivered by aMeta which you perhaps would also like to check:
- the deamination profile computed by MaltExtract among the eight validation and authentication metrics above might be less informative than the one delivered by MapDamage. You can find the deamination profile for the microbe of interest with
taxIDdetected in samplesampleIDhereresults/MAPDAMAGE/sampleID/taxID.tax.bam/Fragmisincorporation_plot.pdf. Please note that the MapDamage deamination profile is computed on Bowtie2 alignments without LCA, these alignments might be less accurate than the LCA-based Malt alignments used for MaltExtract.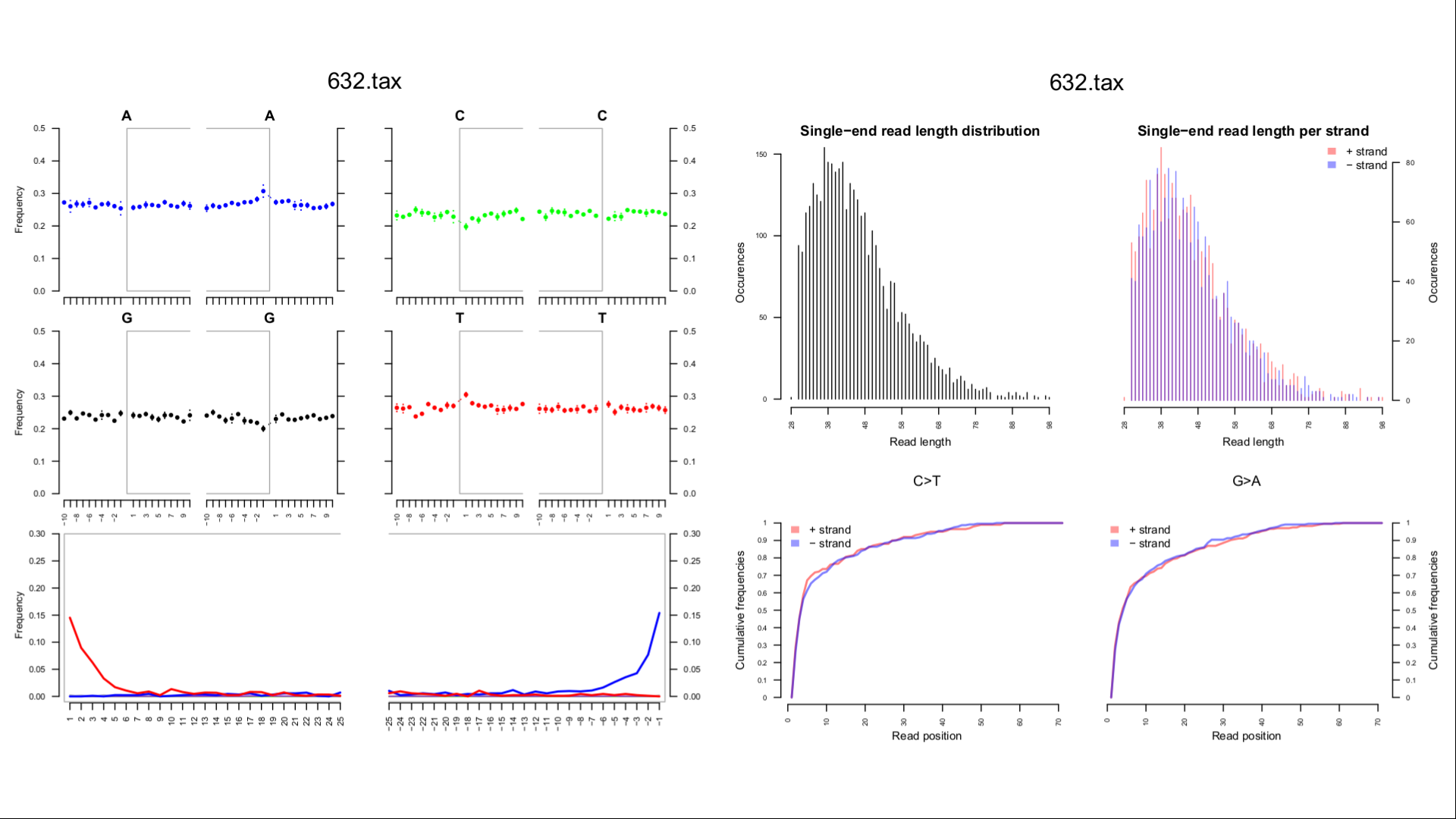
- visualization of KrakenUniq microbial detection results for each sample
sampleIDis available atresults/KRAKENUNIQ/sampleID/taxonomy.krona.html.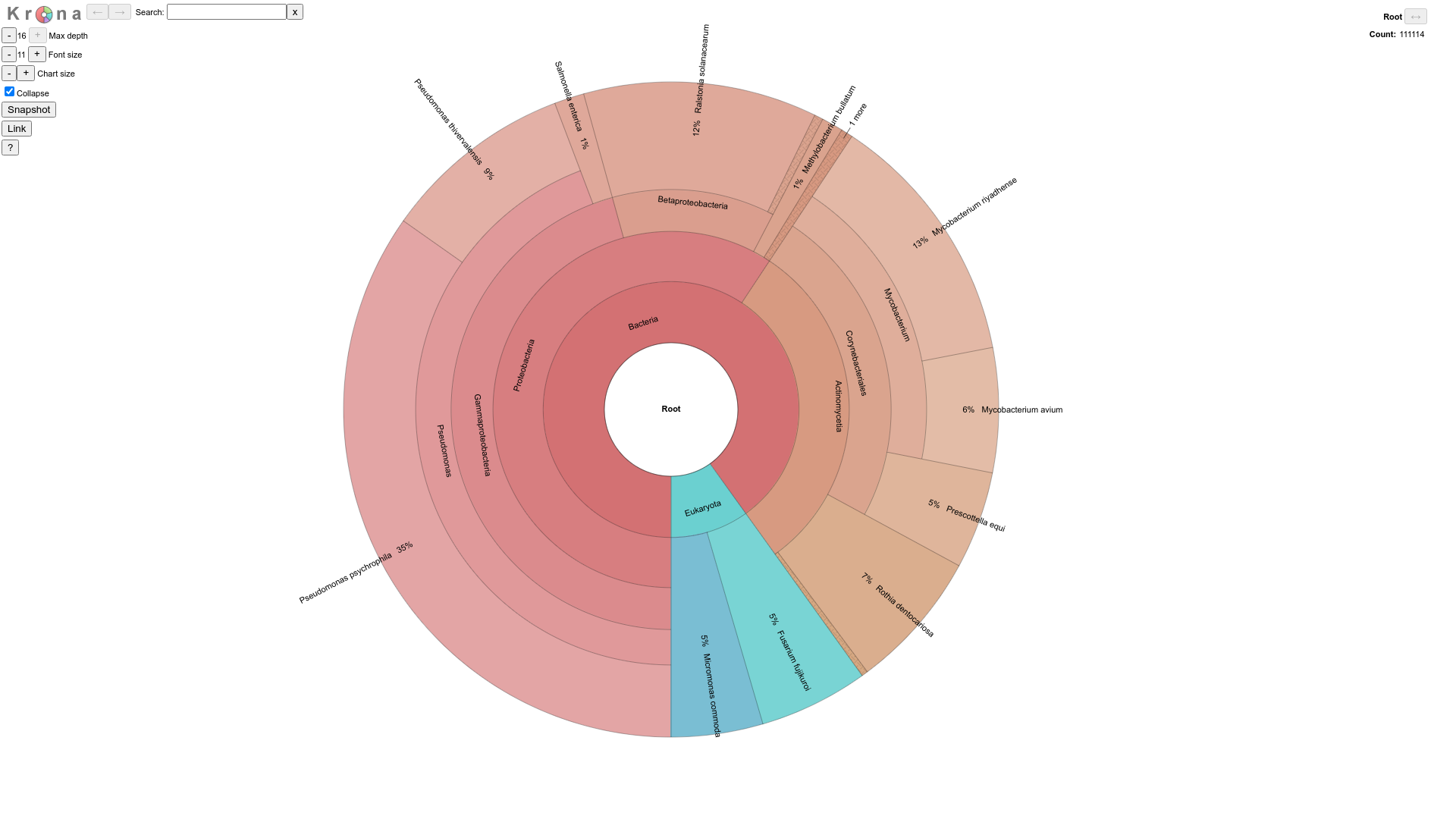
- unfiltered KrakenUniq results for each sample
sampleIDcan be found inresults/KRAKENUNIQ/sampleID/krakenuniq.output, filtered withn_unique_kmers(breadth of coverage) andn_tax_reads(depth of coverage) can be found inresults/KRAKENUNIQ/sampleID/krakenuniq.output.filtered, and the pathogenic subset of the filtered KrakenUniq results for each samplesampleIDis available inresults/KRAKENUNIQ/sampleID/krakenuniq.output.pathogens. - Malt microbial abundance matrix quantified from rma6- and sam-files can be found in
results/MALT_ABUNDANCE_MATRIX_RMA6/malt_abundance_matrix_rma6.txtandresults/MALT_ABUNDANCE_MATRIX_SAM/malt_abundance_matrix_sam.txt, respectively. These abundance matrices are complementary and can be compared with the KrakenUniq abundance matrix fromresults/KRAKENUNIQ_ABUNDANCE_MATRIX/krakenuniq_abundance_matrix.txtfor a better intuition about microbial presence and abundance. - all technical details on the quality of your data, adapter trimming, alignments etc. can be found in
results/MULTIQC/multiqc_report.html.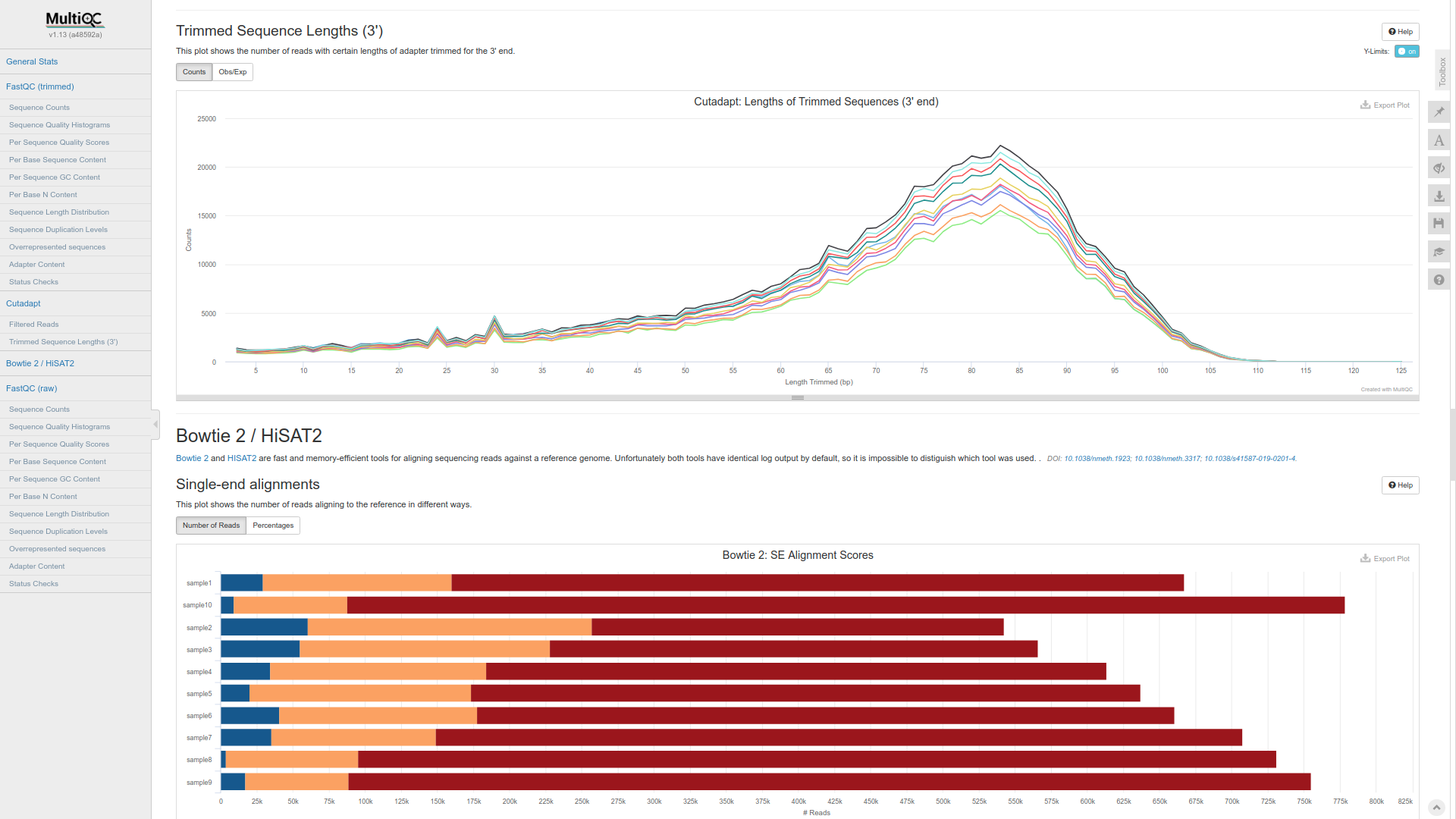



Within config.yaml one can specify what samples to analyse in the samples section through the include and exclude keys, so that a global samplesheet can be reused multiple times.
Analyses mapdamage, authentication, malt, and krona can be individually turned on and off in the analyses section.
Adapter sequences can be defined in the adapters section of config.yaml.
The keys config['adapters']['illumina'] (default true) and config['adapters']['nextera'] (default false) are switches
that turn on/off adapter trimming of illumina (AGATCGGAAGAG) and nextera (AGATCGGAAGAG) adapter sequences. Addional custom adapter sequences can be set in the configuration key
config['adapters']['custom'] which must be an array of strings.
An example snippet that can optionally be added to the configuration file config.yaml is shown below:
# you can include or exclude samples
samples:
include:
- foo
exclude:
- bar
# you can include or exclude certain types of analysis
analyses:
mapdamage: true
authentication: true
malt: true
krona: true
# you can specify type of adapters to trim
adapters:
illumina: true
nextera: false
# custom is a list of adapter sequences
custom: []
To run the workflow in a computer cluster environemnt you should specify environmental modules and runtimes via --profile as follows:
snakemake --snakefile workflow/Snakefile -j 100 --profile .profile --use-envmodules
If the workflow is run on a HPC with the --use-envmodules option
(see
using-environment-modules),
the workflow will check for an additional file that configures environment modules. By default, the file is config/envmodules.yaml, but a custom location can be set with the
environment variable ANCIENT_MICROBIOME_ENVMODULES.
Environmental modules configurations are placed in a configuration section
envmodules with key-value pairs that map a dependency set to a list
of environment modules. The dependency sets are named after the rule's
corresponding conda environment file, such that a dependency set may
affect multiple rules. For instance, the following example shows how
to define modules for rules depending on fastqc, as it would be
implemented on the uppmax compute cluster:
envmodules:
fastqc:
- bioinfo-tools
- FastQC
See the configuration schema file
(workflows/schema/config.schema.yaml) for more information.
Most individual rules define the number of threads to run. Although
the number of threads for a given rule can be tweaked on the command
line via the option --set-threads, it is advisable to put all
runtime configurations in a
profile.
At its simplest, a profile is a directory (e.g. .profile in the
working directory) containing a file config.yaml which consists of
command line option settings. In addition to customizing threads, it
enables the customization of resources, such as runtime and memory. An
example is shown here:
# Rerun incomplete jobs
rerun-incomplete: true
# Restart jobs once on failure
restart-times: 1
# Set threads for mapping and fastqc
set-threads:
- Bowtie2_Alignment=10
- FastQC_BeforeTrimming=5
# Set resources (runtime in minutes, memory in mb) for malt
set-resources:
- Malt:runtime=7200
- Malt:mem_mb=512000
# Set defalt resources that apply to all rules
default-resources:
- runtime=120
- mem_mb=16000
- disk_mb=1000000
For more advanced profiles for different hpc systems, see Snakemake-Profiles github page.
To our experinece, there are very often adapter traces left even after an adapter removing software has been applied to the fastq-files. Therefore, we strongly recommend not to skip the adapter removal step. This step is typically not time consuming and can only be beneficial for the analysis. Otherwise, adapter contamination can lead to severe biases in microbial discovery.
Currently not, but this option will be added soon. As in other aDNA analyses, PE reads need to be merged prior to using aMeta, this can be achieve by e.g. fastp https://github.com/OpenGene/fastp. Alternatively, simple concatenation of R1 and R2 reads as
cat R1.fastq.gz R2.fastq.gz > merged.fastq.gz
is also possible, and the resulting file merged.fastq.gz can be used as input for aMeta.
You will need to adjust Malt max memory usage (64 GB by default) via modifying malt-build.vmoptions and malt-run.vmoptions files.
To locate these files you have to find a Malt conda environment, activate it and replace the default 64 GB with the amount of RAM available on you computer node, in the example below it is 512 GB:
cd aMeta
env=$(grep hops .snakemake/conda/*yaml | awk '{print $1}' | sed -e "s/.yaml://g" | head -1)
conda activate $env
version=$(conda list malt --json | grep version | sed -e "s/\"//g" | awk '{print $2}')
cd $env/opt/malt-$version
sed -i -e "s/-Xmx64G/-Xmx512G/" malt-build.vmoptions
sed -i -e "s/-Xmx64G/-Xmx512G/" malt-run.vmoptions
cd -
conda deactivate
Similarly to Malt, see above, you will need to modify the default memory usage of FastQC. An example of how this can be done is demonstrated below:
cd aMeta
env=$(grep fastqc .snakemake/conda/*yaml | awk '{print $1}' | sed -e "s/.yaml://g" | head -1)
conda activate $env
version=$(conda list fastqc --json | grep version | sed -e "s/\"//g" | awk '{print $2}')
cd $env/opt/fastqc-$version
sed -i -e "s/-Xmx250m/-Xmx10g/" fastqc
cd -
conda deactivate
You are probably running aMeta (or at least some jobs from aMeta) on nodes without internet connection. If this is the case, you have to manually download and provide NCBI taxonomy for MaltExtract, i.e. ncbi.map and ncbi.tre files, from here https://github.com/husonlab/megan-ce/tree/master/src/megan/resources/files and place them to resources/ncbi.
Short answer: no, you do not need to be worried about purple snakemake warning text. Only red messages indicate an error and should be investigated.
If you run aMeta using our pre-built database:
KrakenUniq DB on full NCBI NT: https://doi.org/10.17044/scilifelab.20205504
KrakenUniq DB on microbial part of NCBI NT: https://doi.org/10.17044/scilifelab.20518251
KrakenUniq DB on microbial part of NCBI RefSeq: https://doi.org/10.17044/scilifelab.21299541
Bowtie2 index for full NCBI NT database: https://doi.org/10.17044/scilifelab.21070063
Bowtie2 index on pathogenic microbes of NCBI NT: https://doi.org/10.17044/scilifelab.21185887
it can be very fast (a few hours for a sample with ~10 mln reads) if you have enough RAM (recommended minimum ~200 GB, ideally ~1 TB). Otherwise, runing aMeta with smaller RAM is also possible but results in much longer computation times. We prioritize using large databases for more accurate metagenomic analysis. Alternatively, smaller databases can also be used which might speed up aMeta considerably, but very likely result in less accurate analysis (lower sensitivity and specificity).