Comparing environmental 16S sequences to sequences from isolates.
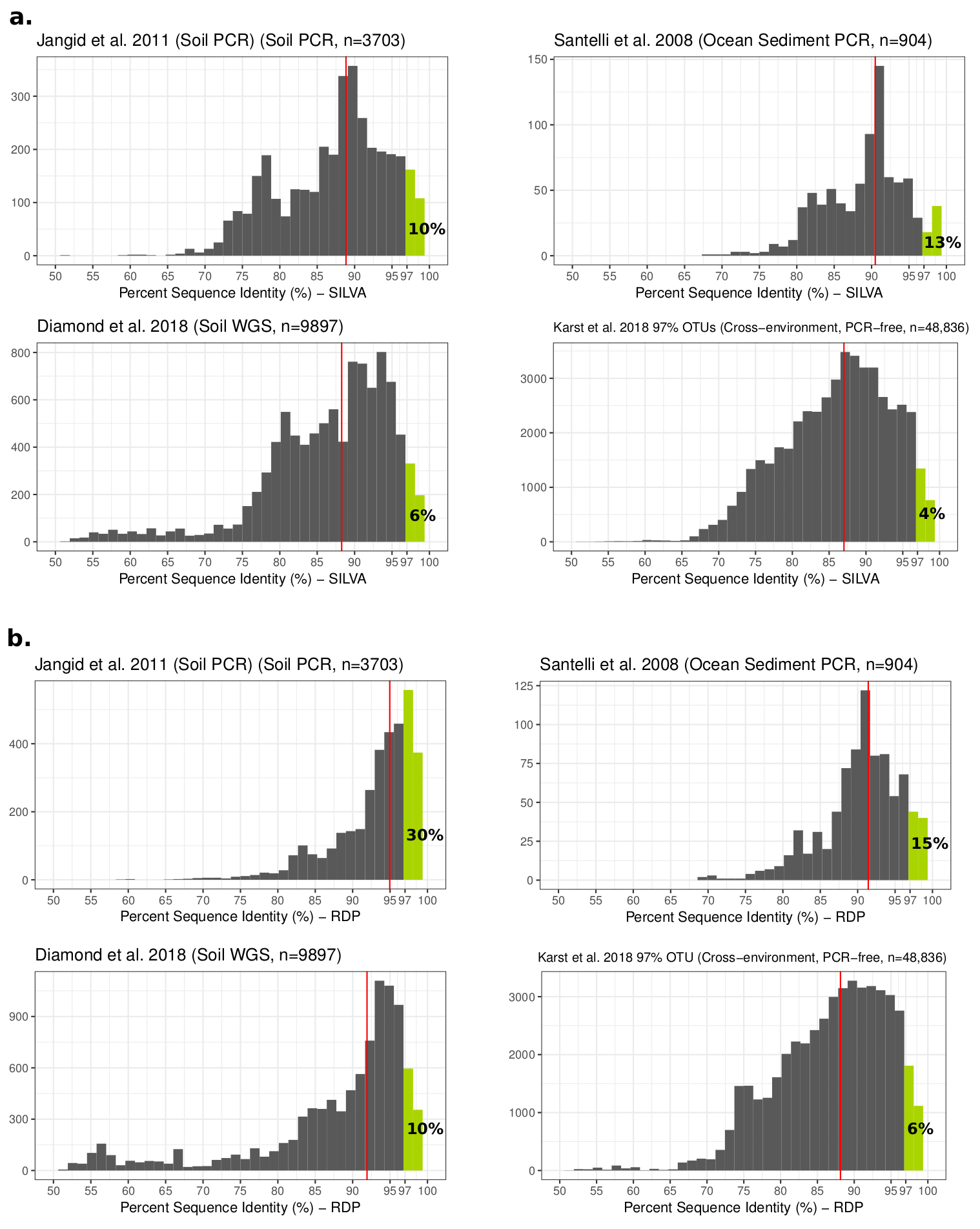
To replicate this analysis:
SILVA: "To get all cultivated strains (including type strains) combine the search for [T] and s[C]" Go to the search page: https://www.arb-silva.de/search/ type [T] into the strain box, search, and add to cart. type [C] into the strain box, search, and add to cart. Download the complete aligment.
RDP:
wget https://rdp.cme.msu.edu/download/current_Bacteria_aligned.fa.gz
gzip -d current_Bacteria_aligned.fa.gz
python filter_rdp_align.py > rdp_filtered.fasta
wget ftp://ftp.ebi.ac.uk/pub/databases/ena/wgs/public/ob/OBAT01.fasta.gz
gzip -d OBAT01.fasta.gz
mothur
align.seqs(candidate=./env_seqs/Jangid2011.16S,template=silva_isolate.fasta)
align.seqs(candidate=./env_seqs/Santelli2008_750bp.16S,template=silva_isolate.fasta)
align.seqs(candidate=./env_seqs/angelo_750bp.16S,template=silva_isolate.fasta)
align.seqs(candidate=./env_seqs/OBAT01.fasta,template=silva_isolate.fasta)
align.seqs(candidate=./env_seqs/Jangid2011.16S,template=rdp_filtered.fasta)
align.seqs(candidate=./env_seqs/Santelli2008_750bp.16S,template=rdp_filtered.fasta)
align.seqs(candidate=./env_seqs/angelo_750bp.16S,template=rdp_filtered.fasta)
align.seqs(candidate=./env_seqs/OBAT01.fasta,template=rdp_filtered.fasta)
The output report files for these commands are included in the mothur_* directories. The scripts Plot.R in each directory recreate the figure above.