


GAUCHE is a collaborative, open-source software library that aims to make state-of-the-art probabilistic modelling and black-box optimisation techniques more easily accessible to scientific experts in chemistry, materials science and beyond. We provide 30+ bespoke kernels for molecules, chemical reactions and proteins and illustrate how they can be used for Gaussian processes and Bayesian optimisation in 10+ easy-to-adapt tutorial notebooks.
Overview | Getting Started | Documentation | Paper (NeurIPS 2023)
- GAUCHE has been accepted to the NeurIPS 2023 Main Track! More details forthcoming!
- Check out our new Molecular Preference Learning and Preferential Bayesian Optimisation notebooks that show how you can use GAUCHE to learn the latent utility function of a human medicinal chemist from pairwise preference feedback!
- Check out our new Sparse GP Regression for Big Molecular Data notebook that shows how you can scale molecular GPs to thousands of data points with sparse inducing point kernels!
General-purpose Gaussian process (GP) and Bayesian optimisation (BO) libraries do not cater for molecular representations. Likewise, general-purpose molecular machine learning libraries do not consider GPs and BO. To bridge this gap, GAUCHE provides a modular, robust and easy-to-use framework of 30+ parallelisable and batch-GP-compatible implementations of string, fingerprint and graph kernels that operate on a range of widely-used molecular representations.
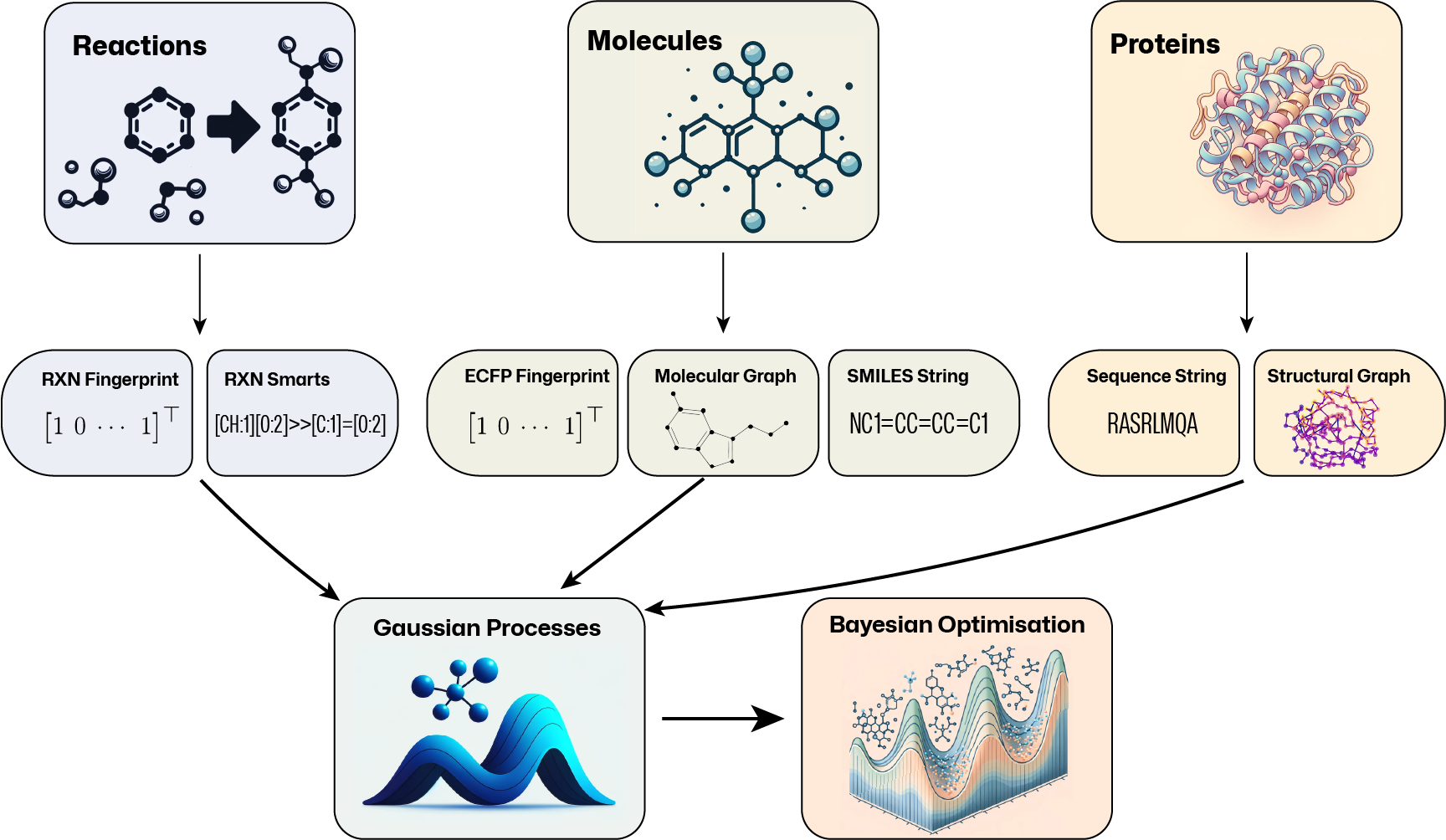
Standard GP packages typically assume continuous input spaces of low and fixed dimensionality. This makes it difficult to apply them to common molecular representations: molecular graphs are discrete objects, SMILES strings vary in length and topological fingerprints tend to be high-dimensional and sparse. To bridge this gap, GAUCHE provides:
- Fingerprint Kernels that measure the similarity between bit/count vectors of descriptor by examining the degree to which their elements overlap.
- String Kernels that measure the similarity between strings by examining the degree to which their sub-strings overlap.
- Graph Kernels that measure between graphs by examining the degree to which certain substructural motifs overlap.
GAUCHE supports any representation that is based on bit/count vectors, strings or graphs. For rapid prototyping and benchmarking, we also provide a range of standard featurisation techniques for molecules, chemical reactions and proteins:
| Domain | Representation |
|---|---|
| Molecules | ECFP Fingerprints [1], rdkit Fragments, Fragprints, Molecular Graphs [2], SMILES [3], SELFIES [4] |
| Chemical Reactions | One-Hot Encoding, Data-Driven Reaction Fingerprints [5], Differential Reaction Fingerprints [6], Reaction SMARTS |
| Proteins | Sequences, Graphs [7] |
If there are any specific kernels or representations that you would like to see included in GAUCHE, feel free to submit an issue or pull request.
The easiest way to install Gauche is via pip.
pip install gaucheAs not all users will need the full functionality of the package, we provide a range of installation options:
pip install gauche- installs the core functionality of GAUCHE (kernels, representations, data loaders, etc.) and should cover a wide range of use cases.pip install gauche[rxn]- additionally installs the rxnfp and drfp fingerprints that can be used to represent chemical reactions.pip install gauche[graphs]- installs all dependencies for graph kernels and representations.
If you aren't sure which installation option is right for you, you can simply install all of them with pip install gauche[all].
The best way to get started with GAUCHE is to check out our tutorial notebooks. These notebooks provide a step-by-step introduction to the core functionality of GAUCHE and illustrate how it can be used to solve a range of common problems in molecular property prediction and optimisation. To install gauche in the colab environment run:
pip install gauche
GAUCHE provides a range of helper functions for loading and preprocessing datasets for molecular property and reaction yield prediction and optimisation tasks. For more detail, check out our Loading and Featurising Molecules Tutorial and the corresponding section in the Docs.
from gauche.dataloader import MolPropLoader
loader = MolPropLoader()
# load one of the included benchmarks
loader.load_benchmark("Photoswitch")
# or a custom dataset
loader.read_csv(path="data.csv", smiles_column="smiles", label_column="y")
# and quickly featurise the provided molecules
loader.featurize('ecfp_fragprints')
X, y = loader.features, loader.labelsFitting a GP model with a kernel from GAUCHE and using it to predict the properties of new molecules is as easy as this. For more detail, check out our GP Regression on Molecules Tutorial and the corresponding section in the Docs.
import gpytorch
from botorch import fit_gpytorch_model
from gauche.kernels.fingerprint_kernels.tanimoto_kernel import TanimotoKernel
# define GP model with Tanimoto kernel
class TanimotoGP(gpytorch.models.ExactGP):
def __init__(self, train_x, train_y, likelihood):
super(TanimotoGP, self).__init__(train_x, train_y, likelihood)
self.mean_module = gpytorch.means.ConstantMean()
self.covar_module = gpytorch.kernels.ScaleKernel(TanimotoKernel())
def forward(self, x):
mean_x = self.mean_module(x)
covar_x = self.covar_module(x)
return gpytorch.distributions.MultivariateNormal(mean_x, covar_x)
# initialise GP likelihood, model and
# marginal log likelihood objective
likelihood = gpytorch.likelihoods.GaussianLikelihood()
model = TanimotoGP(X_train, y_train, likelihood)
mll = gpytorch.mlls.ExactMarginalLogLikelihood(likelihood, model)
# fit GP with BoTorch in order to use
# the LBFGS-B optimiser (recommended)
fit_gpytorch_model(mll)
# use the trained GP to get predictions and
# uncertainty estimates for new molecules
model.eval()
likelihood.eval()
preds = model(X_test)
pred_means, pred_vars = preds.mean, preds.varianceIf GAUCHE is useful for your work please consider citing the following paper:
@article{griffiths2024gauche,
title={{GAUCHE}: A library for {Gaussian} processes in chemistry},
author={Griffiths, Ryan-Rhys and Klarner, Leo and Moss, Henry and Ravuri, Aditya and Truong, Sang and Du, Yuanqi and Stanton, Samuel and Tom, Gary and Rankovic, Bojana and Jamasb, Arian and others},
journal={Advances in Neural Information Processing Systems},
volume={36},
year={2024}
}
[1] Rogers, D. and Hahn, M., 2010. Extended-connectivity fingerprints. Journal of Chemical Information and Modeling, 50(5), pp.742-754.
[2] Fey, M., & Lenssen, J. E. (2019). Fast graph representation learning with PyTorch Geometric. arXiv preprint arXiv:1903.02428.
[3] Weininger, D., 1988. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of Chemical Information and Computer Sciences, 28(1), pp.31-36.
[4] Krenn, M., Häse, F., Nigam, A., Friederich, P. and Aspuru-Guzik, A., 2020. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Machine Learning: Science and Technology, 1(4), p.045024.
[5] Probst, D., Schwaller, P. and Reymond, J.L., 2022. Reaction classification and yield prediction using the differential reaction fingerprint DRFP. Digital Discovery, 1(2), pp.91-97.
[6] Schwaller, P., Probst, D., Vaucher, A.C., Nair, V.H., Kreutter, D., Laino, T. and Reymond, J.L., 2021. Mapping the space of chemical reactions using attention-based neural networks. Nature Machine Intelligence, 3(2), pp.144-152.
[7] Jamasb, A., Viñas Torné, R., Ma, E., Du, Y., Harris, C., Huang, K., Hall, D., Lió, P. and Blundell, T., 2022. Graphein-a Python library for geometric deep learning and network analysis on biomolecular structures and interaction networks. Advances in Neural Information Processing Systems, 35, pp.27153-27167.