This repository contains the code and analyses associated with a single-cell RNAseq study of homeostatic Hydra presented in the following manuscript:
Siebert S, Farrell JA, Cazet JF, Abeykoon Y, Primack AS, Schnitzler CE, Juliano CE (2019) Stem cell differentiation trajectories in Hydra resolved at single-cell resolution. Science 365, eaav9314. https://doi.org/10.1126/science.aav9314
This work was initially presented in the following bioRxiv preprint: https://doi.org/10.1101/460154
The preprint analysis is available as release “Preprint v1” posted on November 3, 2018.
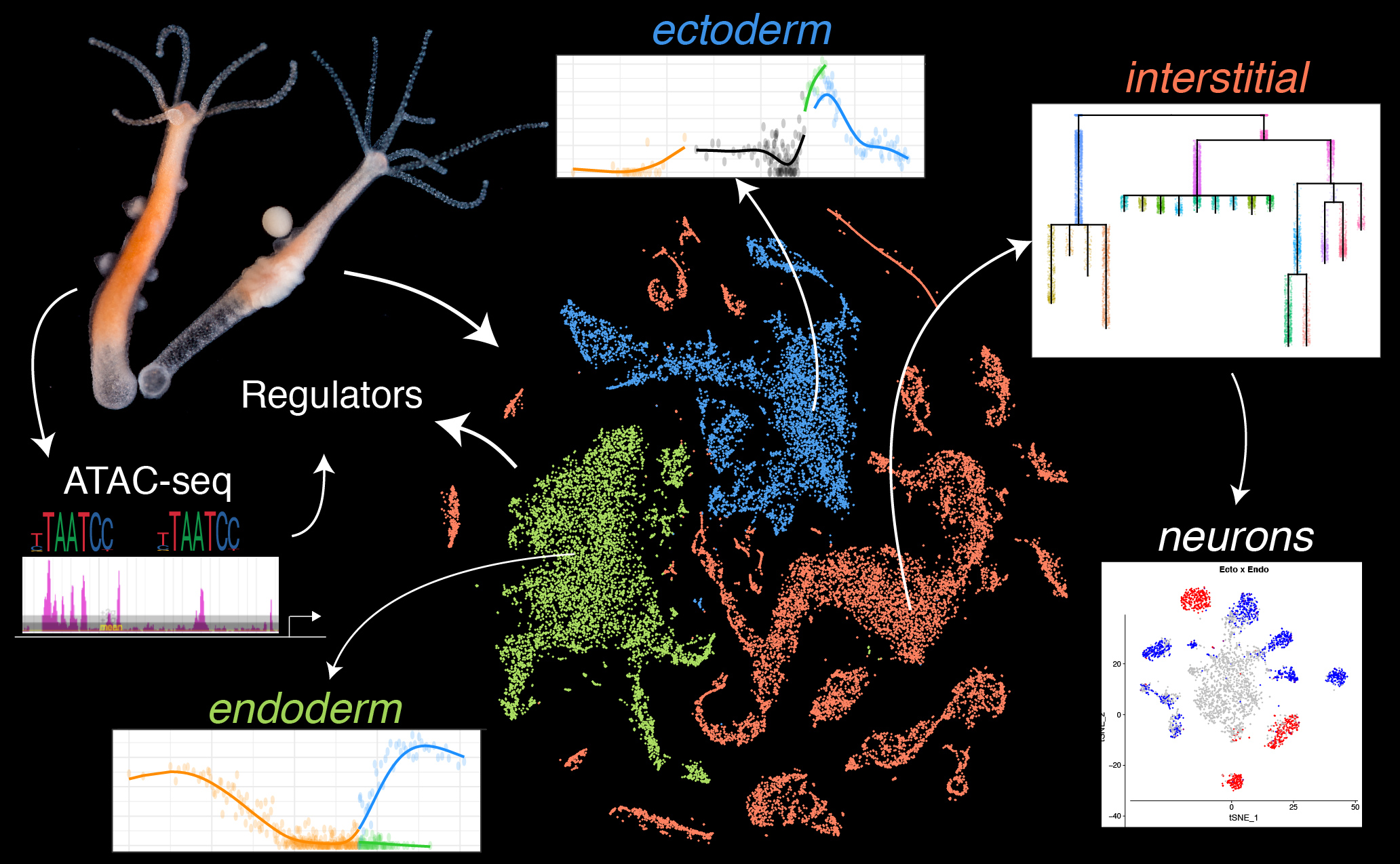
The repository includes a number of files needed to recreate these analyses. UMI count matrices (for transcriptome and genome mapped Drop-seq reads) and isoform level expression estimates for epithelium specific gene expression can be downloaded at the GEO repository; accession GSE121617.
The transcriptome and the genome can be accessed, searched via blast and downloaded at https://research.nhgri.nih.gov/hydra/sequenceserver/. It is also available as Transcriptome Shotgun Assembly project: GHHG00000000, version, GHHG01000000.
ATAC-seq data are available as tracks at https://research.nhgri.nih.gov/hydra/.
Raw RNAseq data used for de novo transcriptome assembly are accessible at SRA; Bioproject: PRJNA497966.
Single cell data are available in a browsable format at:
Selected/final R analysis objects are available from the single-cell portal and from the Dryad Digital Repository: https://doi.org/10.5061/dryad.v5r6077.
R Markdown documents with analysis code (also available as knitted pdfs).
SA01_ClustTranscriptomePermissive.Rmd
- Initial clustering, gene/UMI cut-off decision
SA02_ClustTranscriptome.Rmd
- Clustering final cut-offs (transcriptome)
SA03_SubclustEpithelialCells.Rmd
- Subclusterings for epithelial cells
SA04_SubclustInterstitialCells.Rmd
- Subclustering for cells from the interstitial lineage
SA05_SubclustNeuronalCells.Rmd
- Subclustering for neuronal cells, cell placement
SA06_ClustGenome.Rmd
- Clustering after mapping to Hydra 2.0 genome
SA07_NMF.Rmd - NMF sample analysis (endoderm)
- Non-negative matrix factorization (NMF) analysis, sample analysis (Endodermal epithelial cell subset)
SA08_MotifEnrichmentAnalysis.Rmd
- Motif enrichment analysis, identification of putative regulators
SA09_URD_Endoderm.Rmd
- Trajectory reconstruction for endodermal epithelial cells
SA10_URD_Ectoderm.Rmd
- Trajectory reconstruction for ectodermal epithelial cells
SA11a_URD_InterstitialCellsSubset.Rmd
- Subsetting of cells from the interstitial lineage
SA11b_URD_InterstitialCellsTree.Rmd
- Differentiation tree reconstruction for interstitial cells excluding germline
SA12_URD_GranularZymogen.Rmd
- Trajectory reconstruction for granuluar mucuous and zymogen gland cells
SA13_URD_Spumous.Rmd
- Trajectory reconstruction for spumous mucuous gland cells
SA14_URD_MaleTranscriptome.Rmd
- Trajectory reconstruction for male germline cells (transcriptome data)
SA15_URD_MaleGenome.Rmd
- Trajectory reconstruction for male germline cells (genome data)
The repository also includes the following files:
Plotting Hydra data in URD
- Tutorial showing how to visualize gene expression on trajectories. URD analysis objects are available for download from the Broad Single-cell Portal or from the Dryad Digital Repository: https://doi.org/10.5061/dryad.v5r6077. This tutorial is available as R Markdown document and as knitted html document.
MotifEnrichmentTable.xlsx
- In supplemental table S5 of the manuscript we provide a list of metagenes with associated enriched motifs and candidate key regulators. The content of this table depends on the correlation cutoff used (measure of correlation between expression domains of metagene and putative regulator), with higher cutoffs leading to a more stringent list, but with the tradeoff of possibly missing regulators. Table S5 uses a more stringent correlation cutoff (0.3). We here provide a more extensive list (correlation cutoff of 0.1) for further exploration.
The respository also includes the following folders:
Contains Non-negative matrix factorization (NMF) results for different sets of cells. Provided are cell and gene scores for metagenes with strong cell-type signatures (“good metagenes") and metagenes with more general cell state or technical signature (“bad metagenes”). Also provided are the 30 highest scoring genes for each metagene.
- ec_K76 - NMF for subset of all ectodermal epithelial cells
- ec_K79 - NMF for subset of ectodermal epithelial cells considered in the subcluster analysis (epithelial cell – nematocyte doublets were excluded)
- en_K40 - NMF for subset of all ectodermal epithelial cells
- ic_K75 - NMF for subset of cells from the interstitial lineage
- wg_K84 - NMF for whole dataset (genome mapped reads)
- wt_K96 - NMF for whole dataset (transcriptome mapped reads)
findMotifs_homer.sh- shell script used to run HOMER.2Rep.IDR.mod.bed- ATAC-seq peak consensus file, available as track on the Hydra 2.0 genome browser https://research.nhgri.nih.gov/hydra/.hydra.augustus.nameMod.fastp- Protein sequences derived from Hydra 2.0 gene models used in JASPAR profile inference.hydra.augustus.pfam.filtered.csv- Pfam domains identified in Hydra 2.0 proteins using an independent expect-value equal to or below 1e-6 and with a minimum alignment length of 4aa.motifHeatmapFull.csv- Results table: metagene - identified enriched motif.jaspar2homer.sh- Shell script to reformat JASPAR motifs in folder Hydra_PFMs to HOMER format. Uses:parseJasparMatrix.pl- HOMER script used to convert JASPAR to HOMER format.PWM_Convert.R
TF_domains.txt- List of considered Pfam DBDs. This list was a modified from a previously published set of Pfam domains by adding selected domains (Mendoza et al. 2013, doi:10.1073/pnas.1311818110).Whole_2Rep_IDR_finalhits.txt- File containing peak - gene association (UROPA output, (Kondili et al. 2017, doi: 10.1038/s41598-017-02464-y)S_Enrichment_Workflow.png- Figure to be included in markdown (Fig. 1, SA08_MotifEnrichmentAnalysis.pdf)metaMap.txt- Metagene - cell state annotations. Used as columns in the enrichment matrix that is presented in markdown Fig. 2 (SA08_MotifEnrichmentAnalysis.pdf).Metagene_Gene_Lists/- Contains extended lists of genes that are associated with genome metagenes (wg_K84) and that were considered when identifying regions of open chromatin for subsequent motif enrichment analysis.Hydra_PFMs/- JASPAR motifs identified in Hydra proteins.JASPAR2018_CORE_redundant_pfms_jaspar/- Complete set of available JASPAR motifs (available at http://jaspar.genereg.net)
Many steps of URD involve simulations, which are non-deterministic. Thus, we include the results of our simulations so that results can be reproduced exactly.
ectoderm-flood-dml40-295441.rds- 'Flood' simulations for determining pseudotime in the ectodermal epithelial cellsectoderm-walks-dml40-20F-40B.rds- Biased random walk simulations in the ectodermal epithelial cellsendoderm-flood-dmK60S6-NW-607654556.rds- 'Flood' simulations for determining pseudotime in the endodermal epithelial cellsendoderm-walks-dmK60S6-0F-500B-232119170.rds- Biased random walk simulations in the endodermal epithelial cellsic-clusters.txt- Cluster assignments in the interstitial lineage used for determining the root and tipsic-dm-100NN-localS.rds- Diffusion map in the interstitial lineageic-flood-100NN-localS-39136755.rds- 'Flood' simulations for determining pseudotime in the interstitial lineage cellsic-terminal-nematocytes.txt- List of 'terminal nematocyte' cells that were excluded in building the interstitial lineage differentiation treeic-var.txt- List of variable genes in the interstitial lineage used for determining outlier cellsic-walks-100NN-localS-0F-100B-920281002.rds- Biased random walk simulations in the interstitial cellsmale-flood-dmg75-rootI20_7.rds- 'Flood' simulations for determining pseudotime in the male germline cells (transcriptome aligned, beginning at cluster 7)male-flood-dmg75-rootI20_9.rds- 'Flood' simulations for determining pseudotime in the male germline cells (transcriptome aligned, beginning at cluster 9)malegenome-flood-dmg75-rootI20_12.rds- 'Flood' simulations for determining pseudotime in the male germline cells (genome aligned, beginning at cluster 12)malegenome-flood-dmg75-rootI20_4.rds- 'Flood' simulations for determining pseudotime in the male germline cells (transcriptome aligned, beginning at cluster 4)spumous-flood-dmg75-rootI20_3.rds- 'Flood' simulations for determining pseudotime in the spumous mucous cellszymogen-flood-dmg75-rootI20_10.rds- 'Flood' simulations for determining pseudotime in the granular mucous and zymogen gland cells (beginning at cluster 10)zymogen-flood-dmg75-rootI20_11.rds- 'Flood' simulations for determining pseudotime in the granular mucous and zymogen gland cells (beginning at cluster 11)