In brief, PIC is a program that helps you calculating pausing index from your next-generation sequencing (NGS) samples at functional regions (TSS & Gene body).
RNAPII pausing is a transcription event that RNAPII moves to -30-50bp of TSS and pauses there after engaged. Only when some protein complex like pTEFb come to release the Pausing, RNAPII could elongate to gene body and produce full-length RNA. RNAPII has been implicated as a critical rate-limit step and irreplaceable check point for mRNA transcription.
We developed PIC for easier and more accurate calculation of pausing index. A user can use default or self-defined regions around TSS and gene body to generate the pausing index. Users can use more stringent filtrations, and they can use different modes to output their preferences.
You need Python 2.7, samtools and bedtools to be able to use PIC.
Extract the package or the scripts to the folder containing your data.
-
Set the locations for TSS regions (TSSR) and gene body regions (GBR). Here you can use
extract_genomic_locations.pyto get locations from a gtf file(any unnecessary information like time and version this kinds of tag information should be manually excluded ). This returns 3 outcomes: gene_tss.bed, gene_GB.bed and gene_info.bed. Of course, you can make them bed files by your own .python extract_genomic_locations.py --gtf hg19.gtf --TSSup 50 --TSSdown 300 --GBdown 3000 --gsize ~/Ref/GRCh37/hg19.genomeThe outputs look like this:
gene_tss.bed:
chr1 11820 12168 ENST00000456328.2 . + chr1 11823 12171 ENST00000515242.2 . + chr1 11825 12173 ENST00000518655.2 . +gene_GB.bed
chr1 12168 17408 ENST00000456328.2 . + chr1 12171 17411 ENST00000515242.2 . + chr1 12173 17408 ENST00000518655.2 . +gene_info.bed
chr1 11869 14409 ENST00000456328.2 . + ENSG00000223972.4 DDX11L1 chr1 11872 14412 ENST00000515242.2 . + ENSG00000223972.4 DDX11L1 chr1 11874 14409 ENST00000518655.2 . + ENSG00000223972.4 DDX11L1 -
Many approaches can be used to calculate read counts. We offers you
tag_RPM.pyto get the tag counts from BAM files. Also, other softwares like spp and Homer are also suggested. You can filter your BAM using —-min to get reads with a minimum mapping quality. The script will normalize the read counts by the total number of mapped reads such that we display read counts in RPM (reads per million). This will output 2 files: tag_count.txt and tag_count_normalize.txt.python tag_RPM.py --chip HUVEC_Pol2.bam --input HUVEC_Pol2_input.bam --min 30 --tss gene_tss.bed --gb gene_GB.bedtag_count_normalize.txt: column1 refers to transcript_id, column2-4 refer to location of TSSR, column5-7 refer to location of GBR, column8 refers to strand, column9-12 refer to normalized tag counts of ChIP_TSS, Input_TSS, ChIP_GB, Input_GB.
ENST00000427426.1 chr1 568088 568436 chr1 568436 571817 + 0.125 0.125 71.5 24.75 ENST00000467115.1 chr1 568866 569214 chr1 569214 572120 + 41.375 0.1875 54.5 24.5 ENST00000514057.1 chr1 569027 569375 chr1 569375 572755 + 41.375 0.1875 30.0833333333 24.4375 -
Pausing_Index.pycalculates pausing index based on the table of normalized tag counts. For PI calculation, we subtract normalized ChIP signal in each region against normalized input signal in each region. Then we calculate the pausing index based on the formula. To consolidate PI values from multiple isoforms,--minis used to set a bottom line for the minimum signal at TSSR. You can use--longestto only retain the isoform with longest length, otherwise, the script will choose the isoform with the strongest signal at TSSR.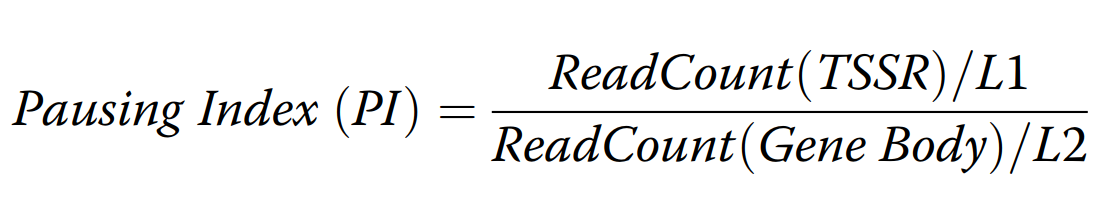
python Pausing_Index.py --tags tag_count_normalize.txt --anno gene_info.txt

Usage: Gene Info extraction [-h] [--gif GTF] [--TSSup TSSUP] [--TSSdown TSSDOWN] [--GBdown GBDOWN] [--gsize GSIZE]
-h, —help show this help message and exit
--gtf GTF the gene annotation file. You should delete titles in this file if exists. Also, please make consistence between the gif and you reference genome. The gtf annotation file can be downloaded from websites like UCSC and GENCODE.
--TSSup TSSUP the distance upstream of TSS
--TSSdown TSSDOWN the distance downstream of TSS
--GBdown GBDOWN the distance downstream of the end of gene body
--gsize GSIZE the chromosome size of the genome, each line: chromosomelength(in bp) . This file can be made by samtools radix on your reference genome and select useful columns using cut option in unix command line.
usage: Calculation RPM for regions [-h] [--chip CHIP] [--input INPUT] [--min MIN] [--tss TSS] [--gb GB]
-h, --help show this help message and exit
--chip CHIP BAM files for ChIP-seq
--input INPUT BAM files for input ChIP-seq
--min MIN Skip alignments with MAPQ smaller than INT[20]
--tss TSS bed file for tss regions
--gb GB bed file for gene body regions
usage: Calculation of Pausing Index [-h] [--tags TAGS] [--min MIN] [--longest] [--anno ANNO]
-h, --help show this help message and exits
--tags TAGS tag counts for each gene
--min MIN minimum ChIP signal at TSSR[0.001]
--longest The script will turn to output the longest isoform if this mode is turned on, otherwise, the script will output the isoform with the strongest signal at TSSR.
--anno ANNO offer gene symbols to gene_id
Chen, J., Y. Fu, D.S. Day, Y. Sun, S. Wang, X. Liang, F. Gu, F. Zhang, S.M. Stevens, P. Zhou, K. Li, Y. Zhang, R.-z. Lin, L.E.H. Smith, J. Zhang, K. Sun, J.M. Melero-Martin, Z. Han, P.J. Park, B. Zhang, and W.T. Pu (2017). "VEGF amplifies transcription through ETS1 acetylation to enable angiogenesis." Nature Communications 8(1): p. 383.
Day, D. S., Zhang, B., Stevens, S. M., Ferrari, F., Larschan, E. N., Park, P. J., & Pu, W. T. (2016). Comprehensive analysis of promoter-proximal RNA polymerase II pausing across mammalian cell types. Genome biology, 17(1), 120.