ICME
A pipeline to identify causal mutations in MutFinderEMS mutant
Recently, I organized the pipeline into a snakemake workflow. Please refer to the link here: https://github.com/xie186/MutFinder/tree/master/snakemake_workflow
Method

WT plants were subjected to EMS mutagenesis. Individuals with desired phenotype in M2 plants were selected to cross with WT plants. Since EMS mutation usually leads to recessive mutants, M2 plants with desired phenotype were considered have homozygous causal mutations.
In the F2 generation, 120 plants with desired phenotype were collected and mixed together for genomic DNA isolation. Re-sequencing reads from wild type and F2 generation were aligned to TAIR10 reference genome using bwa. SAM files generated were then converted to BAM files. PCR duplications were removed using MarkDuplicates.jar in Picard (http://broadinstitute.github.io/picard/). HaplotypeCaller in GATK was then used to identify the SNPs in both wild type and mutant samples. We chose the SNPs meet the following requirements: 1) showing homozygous reference allele in wild type; 2) showing C->T or G->A mutation between wild type and mutant samples; 3) showing ratios of the reads with alternative alleles/read depth > 0.3.
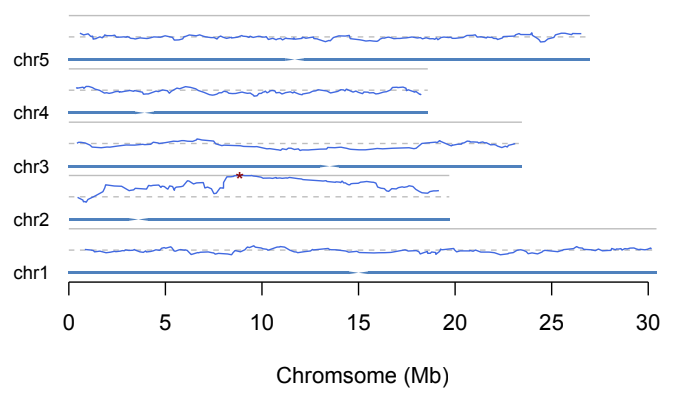
With SNPs meeting the requirements above, a five-snp window was used to calculate the ratio of mutation. Based on the ratios of the windows, a line plot was generated along the chromosome. The peak position was marked with a red star. snpEff (reference) was then used to annotate the effect of the SNPs. SNPs with large effects around the peak position was extracted to identify the causal mutation.
Here is an example:
References:
bwa reference
Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25:1754-60. [PMID: 19451168]
SNPeff
A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff
GATK
The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA, 2010 GENOME RESEARCH 20:1297-303