The purpose of this exercise is to introduce the participants of the Auburn University Bioinformatics Bootcamp to using Git to version control their science!
At this point, you should have (1) an account on Github, (2) been introduced to the very basics of Git, and (3) be working in a group with about 3 other people.
-
All of you should login to your Github account.
-
One of you should fork this repository, by clicking the 'Fork' button on the upper right of the page.
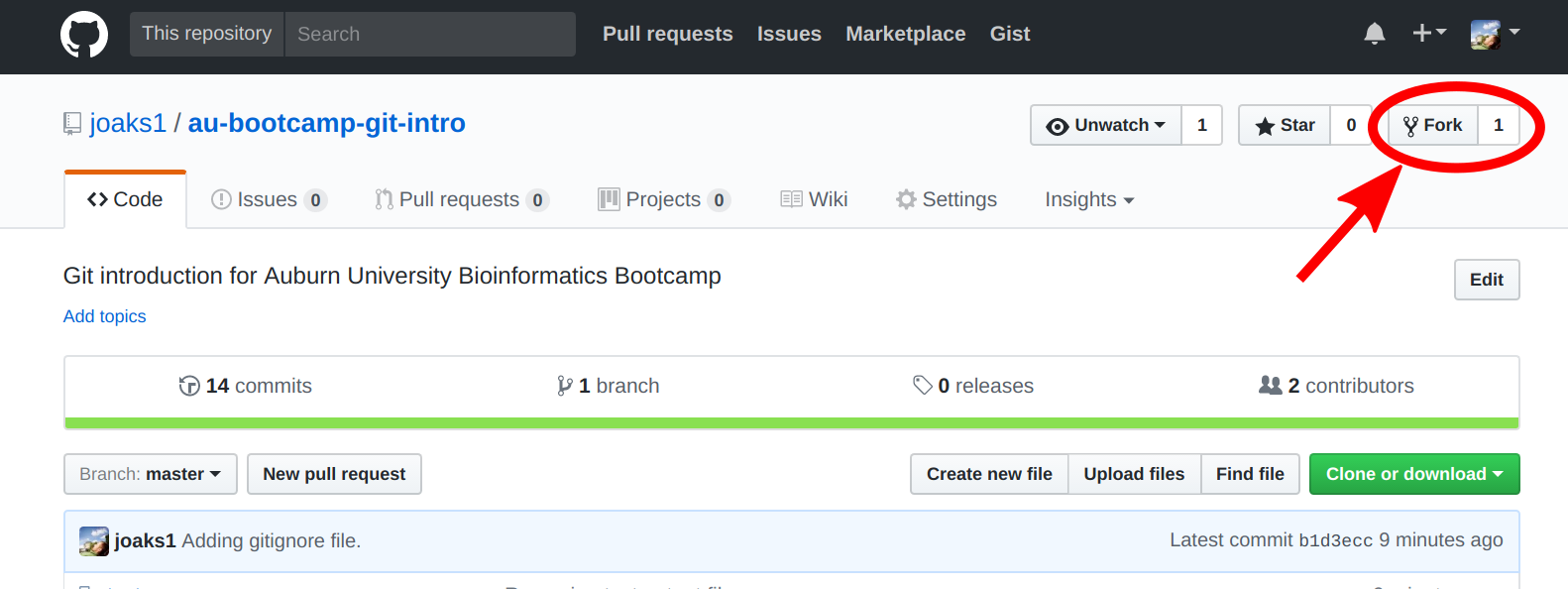
After a few seconds, the team member that forked should be looking at their copy of the repo in their Github account.
-
Next, the person that forked the repo needs to add their team members as collaborators:
-
Click the 'Settings' tab near the top of the page.
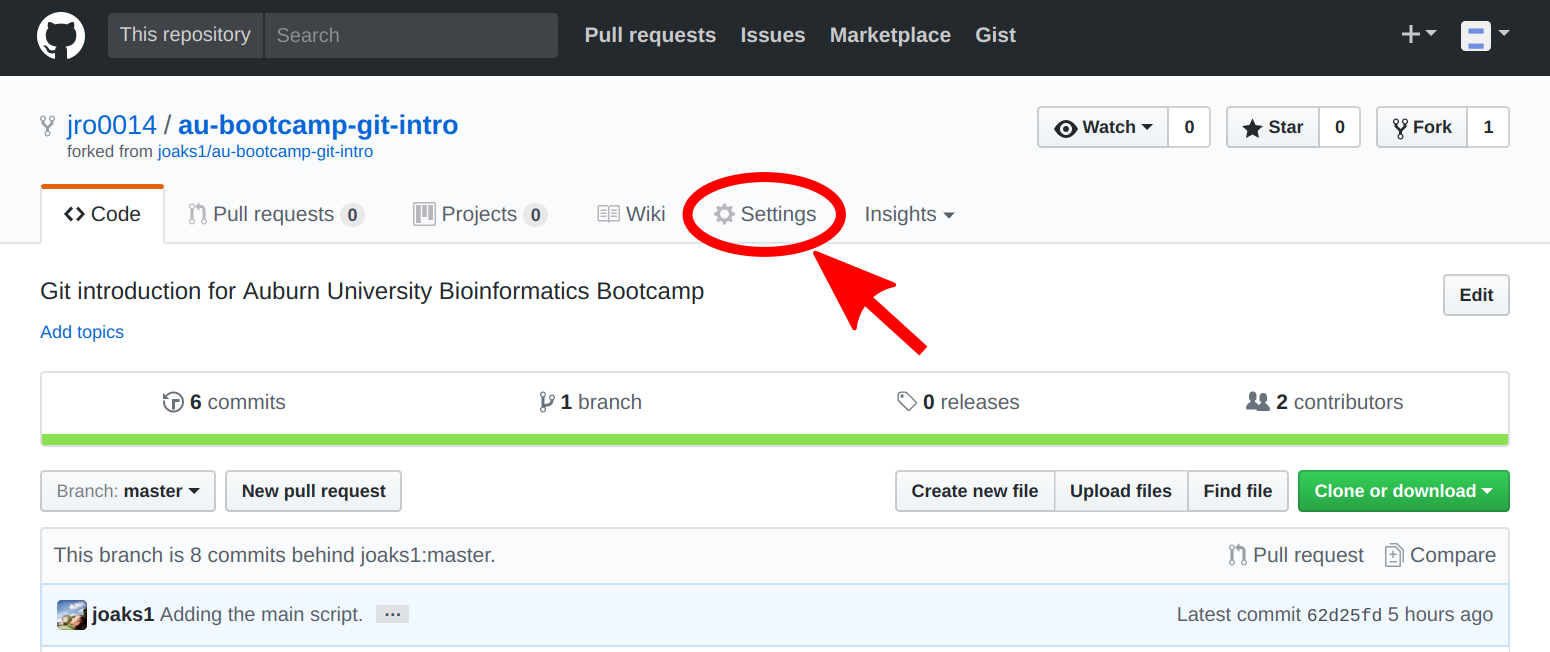
-
Click 'Collaborators' in the settings menu along the left.
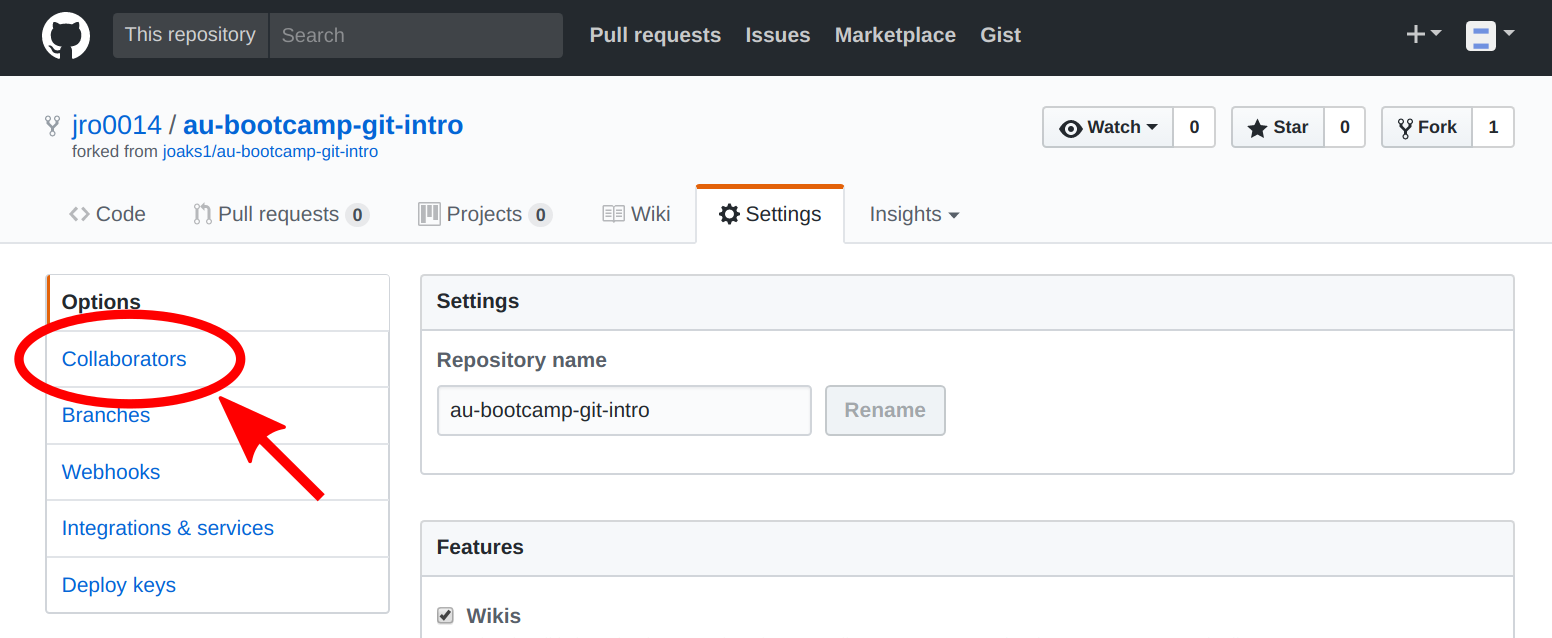
-
You should be able to find your team members' accounts by searching for their Github username.
-
-
Each team member that is added will receive an invitation via e-mail to collaborate on the repo. Accept the invitation and follow the link to the forked repo on your team member's Github account.
-
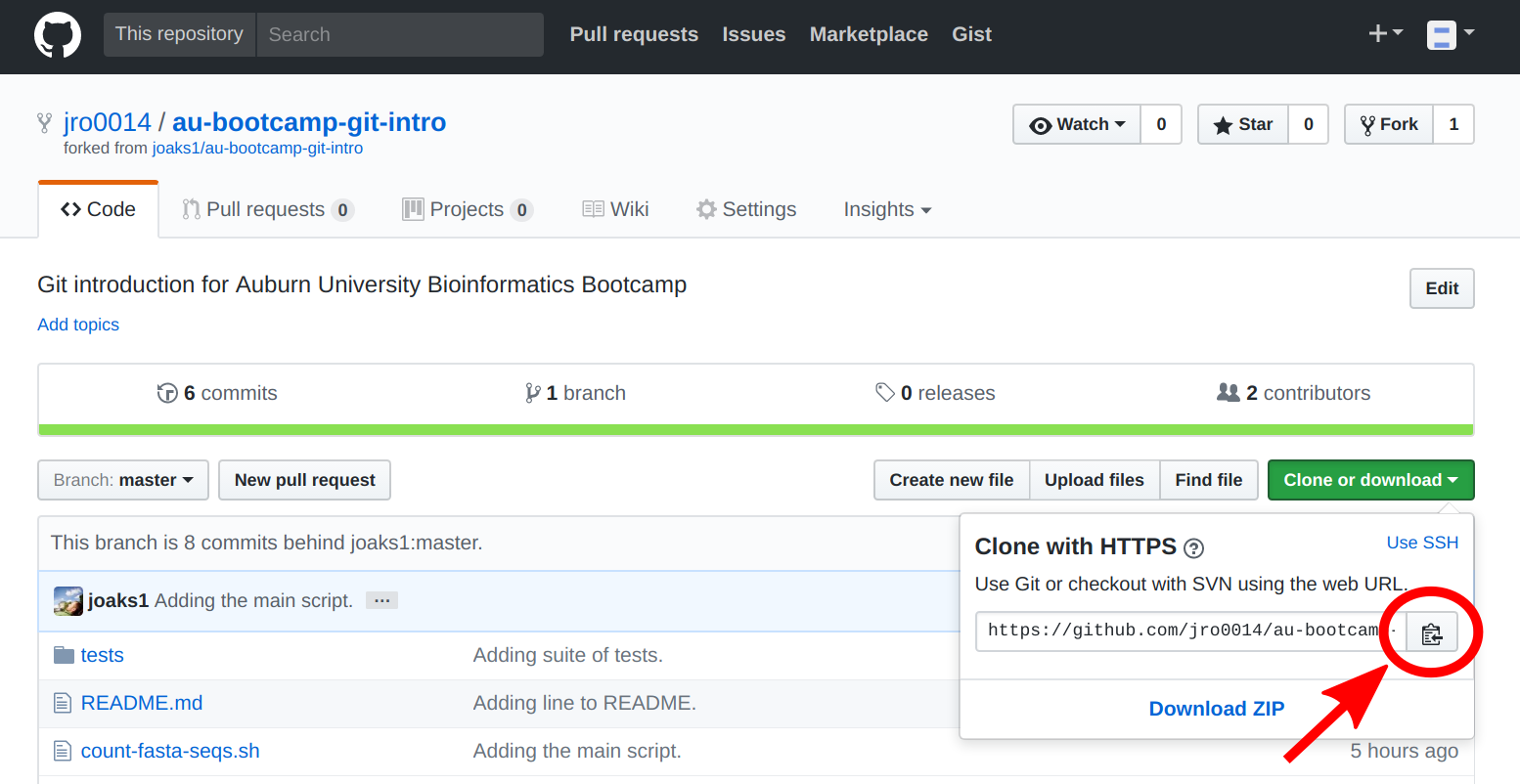
At this point, you should all be on the Github page of your team's forked repo. Make sure you are on the page of your team member's forked repo, NOT my copy of the repo (see image below).
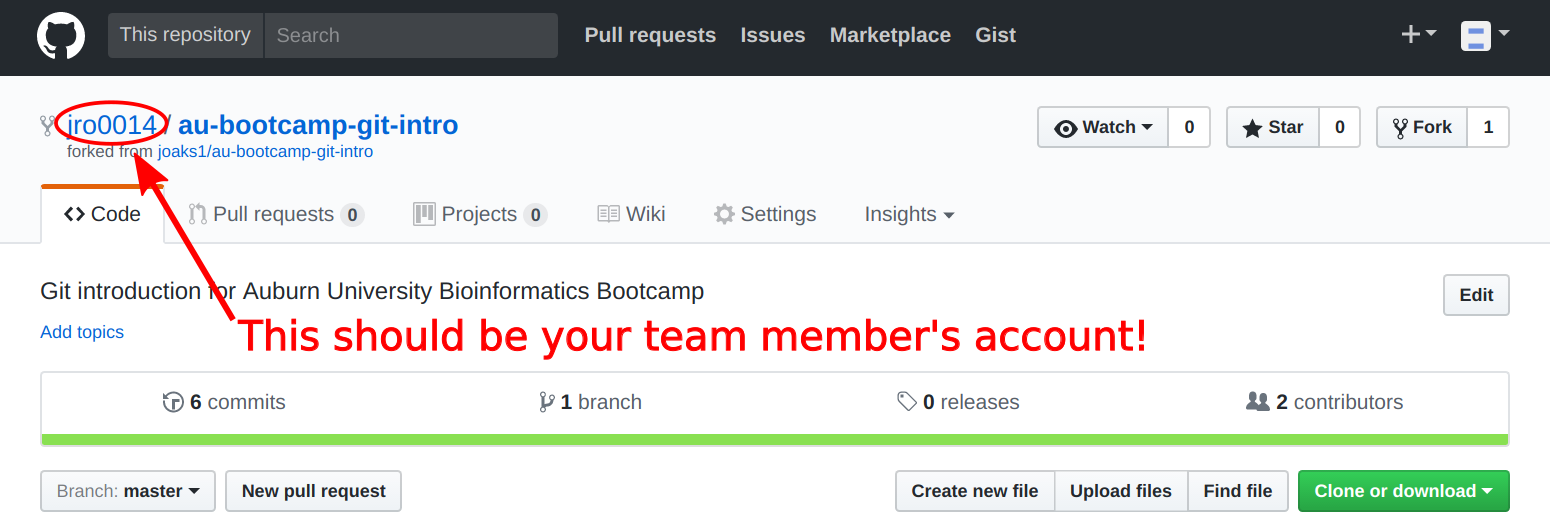
-
Click the 'Clone or download' button, and copy the URL of the repo via the 'copy to clipboard' button. (ALL team members need to do this, including the one who forked the repo)
-
In your terminal, navigate to where you want to keep this repo (you can always move it later, so just your home directory is fine). Then type:
$ git clone <the-url-you-just-copied>and hit enter to clone the repository.
-
Next,
cdinto the directory:$ cd <the-name-of-directory-you-just-cloned> -
At this point, all members of your team should be in their own local copy of the repository.
Each of you now has your very own local copy of the repository, and can
pullandpushchanges from and to the remote copy of the repo hosted on Github.Note, there is nothing special about the remote copy. It is just like the local copy of the repo you have. The only difference is that it is located in a place you can all access (on a Github server). This is what it means when we say Git is distributed; everyone has a full copy of the entire repo!





The goal of this exercise is for you to work collaboratively with your team members to write a simple shell script.
Type ls in the directory of the repo you just cloned. You should see
something like:
$ ls
count-fasta-seqs.sh
example-seqs1.fasta
example-seqs2.fasta
LICENSE.txt
README.md
run_tests.sh
tests
util
The file count-fasta-seqs.sh is where you will write your shell script. Go
ahead and open it with your preferred text editor (probably nano if you are
working on the Alabama Super Computer and have become familiar with the nano
during the Bioinformatics Bootcamp).
You will see that the file is mostly just filled with comments (i.e., there's
no code).
The file run_tests.sh is a script that runs a series of tests on the
count-fasta-seqs.sh script. Since that code doesn't exist yet, all of these
tests currently fail. Try it:
$ sh run_tests.sh
----------------------------------------------------------------------
RUNNING TEST:
sh count-fasta-seqs.sh gekko-mindorensis.fasta
*****************************************
FAIL: Did not create the expected output!
*****************************************
Here is the expected output:
19 gekko-mindorensis.fasta
19
-----------------------------------------
Here is the observed output:
gekko-mindorensis.fasta
*****************************************
Passed 0 out of 1 tests
----------------------------------------------------------------------
----------------------------------------------------------------------
RUNNING TEST:
sh count-fasta-seqs.sh cyrtodactylus-philippinicus.fasta gekko-mindorensis.fasta insulasaurus-arborens.fasta
*****************************************
FAIL: Did not create the expected output!
*****************************************
Here is the expected output:
20 cyrtodactylus-philippinicus.fasta
19 gekko-mindorensis.fasta
32 insulasaurus-arborens.fasta
71
-----------------------------------------
Here is the observed output:
cyrtodactylus-philippinicus.fasta gekko-mindorensis.fasta insulasaurus-arborens.fasta
*****************************************
Passed 0 out of 1 tests
----------------------------------------------------------------------
----------------------------------------------------------------------
RUNNING TEST:
sh count-fasta-seqs.sh /home/jamie/Dropbox/projects/au-bootcamp-git-intro/tests/test_nested_singleton/input/gekko-mindorensis.fasta
*****************************************
FAIL: Did not create the expected output!
*****************************************
Here is the expected output:
19 gekko-mindorensis.fasta
19
-----------------------------------------
Here is the observed output:
/home/jamie/Dropbox/projects/au-bootcamp-git-intro/tests/test_nested_singleton/input/gekko-mindorensis.fasta
*****************************************
Passed 0 out of 1 tests
----------------------------------------------------------------------
----------------------------------------------------------------------
RUNNING TEST:
sh count-fasta-seqs.sh /home/jamie/Dropbox/projects/au-bootcamp-git-intro/tests/test_nested_files/input/*
*****************************************
FAIL: Did not create the expected output!
*****************************************
Here is the expected output:
20 cyrtodactylus-philippinicus.fasta
19 gekko-mindorensis.fasta
32 insulasaurus-arborens.fasta
71
-----------------------------------------
Here is the observed output:
/home/jamie/Dropbox/projects/au-bootcamp-git-intro/tests/test_nested_files/input/cyrtodactylus-philippinicus.fasta /home/jamie/Dropbox/projects/au-bootcamp-git-intro/tests/test_nested_files/input/gekko-mindorensis.fasta /home/jamie/Dropbox/projects/au-bootcamp-git-intro/tests/test_nested_files/input/insulasaurus-arborens.fasta
*****************************************
Passed 0 out of 1 tests
----------------------------------------------------------------------
Some tests failed (see messages above)
Four tests were run and, as expected, they all failed.
Your goal is to work collaboratively with your team members to add shell code
to count-fasta-seq.sh until these tests pass.
NOTE: You do NOT need to know how the tests work! You only need to add code to count-fasta-seq.sh (more on what that entails in the next section!)
Make sure you are working together by sharing (pushing and pulling) your
code as you go via Git.
Now, you need to know what the failing tests expect count-fasta-seqs.sh to be
doing!
Below, is some info about what this script should do, including what the inputs
and output should be:
Paths to one or more fasta sequence files.
For each file, it should write a line with the number of sequences in the file, a space, and then the file NAME (NOT the path!), and a final line with the total number of sequences across all files.
In the same directory as this script, you should find an example fasta file named 'example-seqs1.fasta', which contains:
>RMB3263_Cyrtodactylus_philippinicus_Negros
CGGGCCCATACCCCGAAAATGTTGGTATAAACCCCTTCCTATACTAATAAACCCCATTATTTGATCACTATTACTAAC
>CWL052_Cyrtodactylus_philippinicus_Negros
CGGGCCCATACCCCGAAAATGTTGGTATAAACCCCTTCCTATACTAATAAACCCCATTATTTGATCACTATTACTAAC
If you run this script on this fasta file like this
$ sh count-fasta-seqs.sh example-seqs1.fasta
you want to get the following output:
2 example-seqs1.fasta
2
There should be another example fasta file named 'example-seqs2.fasta', which contains:
>RMB7155_Sphenomorphus_arborens_Negros
ATGAACCCCATTATAACCTCCCTCATTTTATCAAGCCTGGCCCTTGGAACCGTAATCACACTAACAAGCTACCACTGA
>RMB7156_Sphenomorphus_arborens_Negros
ATGAACCCCATTATAACCTCCCTCATTTTATCAAGCCTGGCCCTTGGAACCGTAATCACACTAACAAGCTACCACTGA
>RMB7163_Sphenomorphus_arborens_Negros
ATGAACCCCATTATAACCTCCCTCATTTTATCAAGCCTGGCCCTTGGAACCGTAATCACACTAACAAGCTACCACTGA
If you run this script on BOTH fasta files, you want to get the following output:
$ sh count-fasta-seqs.sh example-seqs1.fasta example-seqs2.fasta
2 example-seqs1.fasta
3 example-seqs2.fasta
5
The first thing you need to be able to do is access the paths to the fasta
files that are 'given to' this script. The variable $@ will be very useful
for this. Currently, the only thing count-fasta-seqs.sh does is:
echo "$@"
What does this do? Try this:
$ sh count-fasta-seqs.sh larry curly moe
What is the output? It's just listing the arguments given to the script! Now try:
$ sh count-fasta-seqs.sh example-seqs1.fasta example-seqs2.fasta
Hmmm... That output seems like it might be useful in your script? Now, you need to figure out how to work with each argument. HINT: for loop (remember "for do done"?). Something like:
for filepath in "$@"
do
YOUR CODE HERE
done
just might get you started.
You will also need to get only the name of each file from the path. Checkout the command 'basename' for this:
$ man basename
To count the number of sequences in each file, I recommend you checkout
grep and wc:
$ man grep
$ man wc
WARNING about grep: ALWAYS quote the string that you are trying to find!
For example, do:
$ grep "string I want to find" file-i-want-to-find-it-in.txt
NOT
$ grep string I want to find file-i-want-to-find-it-in.txt # DON'T DO THIS!
To keep a tally of the total number of sequences across all files, expr
might be useful:
$ man expr
REMEMBER: The goal of this exercise is to use git. Don't forget to commit and pull/push often.
Good luck!
This exercise was inspired by, and borrowed heavily from, the Git exercise written by Mark Holder, which can be found at https://github.com/mtholder/swc-tree-support-ex.
This work was made possible by funding provided to Jamie Oaks from the National Science Foundation (DEB 1656004).
This work is licensed under a Creative Commons Attribution 4.0 International License.
