
-
For researchers, who need to investigate genotype phenotype associations, smmart-g2p is a search tool that aggregates evidence from several knowledge bases unlike ad-hoc searches, the product allows the researcher to focus on the evidence, not on the search. more
-
Quickly determine the diseases, drugs and outcomes based on evidence from trusted sources. Find relevant articles and (soon) drug response data.
-
Inform GA4GH G2P discussions
A prototype of the Genotype to phenotype user interface exists here.
Now:
- Jackson Lab Clinical Knowledge Base
- Washington University CIViC
- oncokb Precision Oncology Knowledge Base
- Cancer Genome Interpreter Cancer bioMarkers database
- GA4GH reference server
- Cornell pmkb
- MolecularMatch
In progress:
To see analysis of harmonization and overlaps see figures
JUST GOOGLE IT:
-
Use the search box like a google search. To search your data, enter your search criteria in the Query bar and press Enter or click Search to submit the request. For a full explanation of the search capabilities see Search examples, syntax
-
The charts and list are all tied to the search. Click to constrain your results
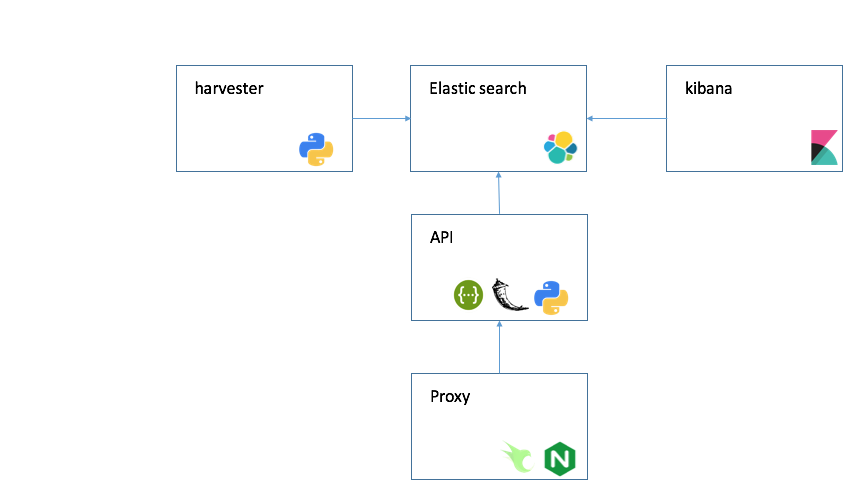
- ElasticSearch, Kibana v6.0
- Python 2.7
- Provisioned by your OS image
- API Flask 0.12.2
- Provisioned by docker
- nginx openresty/openresty
- Provisioned by docker, for more information see cloud setup
On top of Elasticsearch, we built REST-based web services using the Flask web framework.
search.cancervariants.org provides two simple REST-based web services: an association query service and a GA4GH beacon service. The association query service allows users to query for evidence using any combination of keywords, while the beacon service provisions associations into the GA4GH beacon network enabling retrieval of associations based on genomic location.
- Start up an elastic search container
- Register and download CosmicMutantExport.csv into the harvester directory
- Make the required files from the harvester Makefile
$ cd harvester
$ make oncokb_all_actionable_variants.tsv cgi_biomarkers_per_variant.tsv cosmic_lookup_table.tsv cgi_mut_benchmarking.tsv oncokb_mut_benchmarking.tsv benchmark_results.txt
Note: If you will be extracting from molecularmatch, you will need to contact them from an API key. Disease normalization depends on bioontology, see https://bioportal.bioontology.org/accounts/new for an API key.
- Install required python packages
pip install -r requirements.txt
- Run the harvester
$ python harvester.py -h
usage: harvester.py [-h] [--elastic_search ELASTIC_SEARCH]
[--elastic_index ELASTIC_INDEX] [--delete_index]
[--delete_source]
[--harvesters HARVESTERS [HARVESTERS ...]]
optional arguments:
-h, --help show this help message and exit
--elastic_search ELASTIC_SEARCH, -es ELASTIC_SEARCH
elastic search endpoint
--elastic_index ELASTIC_INDEX, -i ELASTIC_INDEX
elastic search index
--delete_index, -d delete elastic search index
--delete_source, -ds delete all content for source before harvest
--harvesters HARVESTERS [HARVESTERS ...]
harvest from these sources. default: ['cgi_biomarkers', 'jax', 'civic', 'oncokb', 'g2p']
A harvester is a python module that implements this duck typing interface.
#!/usr/bin/python
def harvest(genes):
""" given a list of genes, yield an evidence item """
# for gene in genes:
# gene_data = your_implementation_goes_here
# yield gene_data
pass
def convert(gene_data):
""" given a gene_data in it's original form, produce a feature_association """
# gene: a string gene name
# feature: a dict representing a ga4gh feature https://github.com/ga4gh/ga4gh-schemas/blob/master/src/main/proto/ga4gh/sequence_annotations.proto#L30
# association: a dict representing a ga4gh g2p association https://github.com/ga4gh/ga4gh-schemas/blob/master/src/main/proto/ga4gh/genotype_phenotype.proto#L124
#
# feature_association = {'gene': gene ,
# 'feature': feature,
# 'association': association,
# 'source': 'my_source',
# 'my_source': {... original data from source ... }
# yield feature_association
pass
def harvest_and_convert(genes):
""" get data from your source, convert it to ga4gh and return via yield """
for gene_data in harvest(genes):
for feature_association in convert(gene_data):
yield feature_association
$ cd harvester
$ pytest -s -v
======================================================================================================================================================= test session starts ========================================================================================================================================================
platform darwin -- Python 2.7.13, pytest-3.0.7, py-1.4.33, pluggy-0.4.0 -- /usr/local/opt/python/bin/python2.7
cachedir: ../../.cache
rootdir: /Users/walsbr, inifile:
collected 13 items
tests/integration/test_elastic_silo.py::test_args PASSED
tests/integration/test_elastic_silo.py::test_init PASSED
tests/integration/test_elastic_silo.py::test_save PASSED
tests/integration/test_elastic_silo.py::test_delete_all PASSED
tests/integration/test_elastic_silo.py::test_delete_source PASSED
tests/integration/test_kafka_silo.py::test_populate_args PASSED
tests/integration/test_kafka_silo.py::test_init PASSED
tests/integration/test_kafka_silo.py::test_save PASSED
tests/integration/test_pb_deserialize.py::test_civic_pb PASSED
tests/integration/test_pb_deserialize.py::test_jax_pb PASSED
tests/integration/test_pb_deserialize.py::test_oncokb_pb PASSED
tests/integration/test_pb_deserialize.py::test_molecular_match_pb PASSED
tests/integration/test_pb_deserialize.py::test_cgi_pb PASSED
There is a docker compose configuration file in the root directory.
Launch it by:
ELASTIC_PORT=9200 KIBANA_PORT=5601 docker-compose up -d
This will automatically download elastic search etc. and will expose the standard elastic search and kibana ports (9200 and 5601)
If you would like to host an instance, launch docker-compose with an additional nginx file.
docker-compose -f docker-compose.yml -f cloud-setup/docker-compose-nginx.yml up -d
This will do the same setup, but will also include an nginx proxy to map http and https ports.
Our demo site is hosted on aws and includes the API server and nginx proxy
docker-compose -f docker-compose-aws.yml up -d
As a convenience, there is a juypter image for notebook analysis:
docker-compose -f docker-compose.yml -f docker-compose-jupyter.yml up -d
- See the README.md in harvester/tests/integration to see how harvested evidence is mapped to protocol buffer messages.
- Work with users, gather feedback
- Load alternative data sources [literome, ensemble]
- Load smmart drugs [Olaparib, Folfox, Pembrolizumab, …]
- Integrate with bmeg (machine learning evidence)
- Improve data normalization
- Variant naming (HGVS)
- Ontologies (diseases, drugs, variants)
- Add GA4GH::G2P api (or successor)
- Harden prototype:
- python notebook
- web app (deprecate kibana UI)