Functional Mapping and Analysis Pipeline for metagenomics and metatranscriptomics studies
Some example results are available at the homepage: https://qbrc.swmed.edu/FMAP/.
-
FMAP provides a more sensible reference protein sequence database based on UniRef.
-
Identification of differentially-abundant genes KEGG Orthology
-
Mapping differentially-abundant genes to pathways and modules (KEGG Pathway and KEGG Module)
-
Mapping differentially-abundant genes to operons (ODB (v3))
-
Perl - scripting language
-
R - statistical computing
-
Statistics::R - Perl interface with the R statistical program
- Use CPAN to install the module
perl -MCPAN -e 'install Statistics::R'- or download the source and compile manually
wget 'http://search.cpan.org/CPAN/authors/id/F/FA/FANGLY/Statistics-R-0.34.tar.gz' tar zxf Statistics-R-0.33.tar.gz cd Statistics-R-0.33 perl Makefile.PL make make test make install -
Mapping program providing BLASTX search of sequencing reads: DIAMOND or USEARCH
-
Linux commands:
wget,cat,sort -
Bio::DB::Taxonomy - Access to a taxonomy database (which is required only if you want to build a custom database.)
-
XML::LibXML - Perl Binding for libxml2 (which is required only if you want to download genome sequences.)
- FMAP_database.pl
- Process
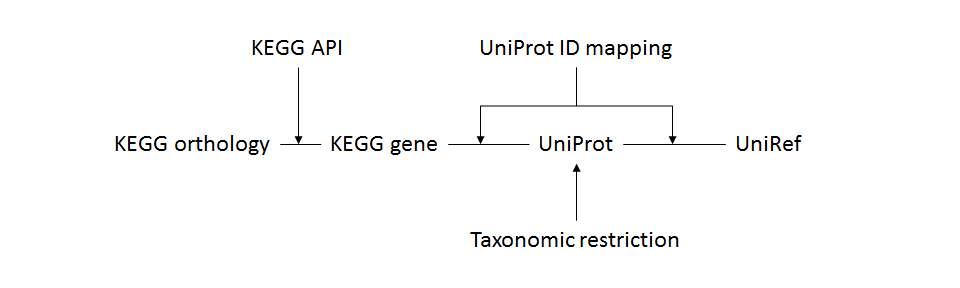
- Input
- UniRef sequence identity (50, 90, or 100)
- (optional) NCBI taxonomy IDs (integer)
- Require Bio::DB::Taxonomy.
- The following data files will be downloaded through FTP connection. If you have a problem in the FTP connection, please download the files through another method and copy them into "FMAP_data" directory before executing "FMAP_database.pl" command.
ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz
ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/idmapping/idmapping.dat.gz
ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref100/uniref100.fasta.gz
or ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref90/uniref90.fasta.gz
or ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref50/uniref50.fasta.gz - Require HTTP connection for KEGG API.
- Process

Usage: perl FMAP_database.pl [options] 50|90|100 [NCBI_TaxID [...]]
Options: -h display this help message
-s switch database
-r redownload data
- FMAP_prepare.pl
Usage: perl FMAP_prepare.pl [options]
Options: -h display this help message
-r redownload data
-m FILE executable file path of mapping program, "diamond" or "usearch" [diamond]
-k download prebuilt KEGG files
- FMAP_assembly.pl
- Process
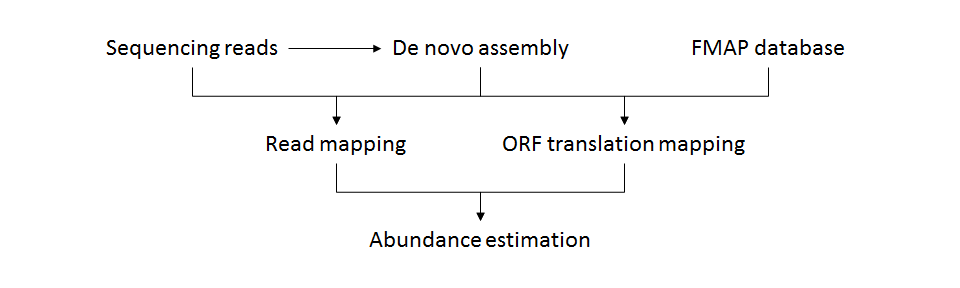
- Input
- Prefix of output files
- De novo assembled sequences in FASTA format
- A FASTA file can be generated by metagenome assemblers such as SPAdes and MetaVelvet.
- A FASTA file containing target genome sequences can be input instead.
- Whole metagenomic/metatranscriptomic shotgun sequencing reads in FASTQ or FASTA format
- Multiple read files can be specified.
- Paired-end read files must be specified comma-separated like "input.R1.fastq,input.R2.fastq".
- The read files can be compressed by gzip.
- Output
- Prefix.region.abundance.txt (abundances of ORF regions mapping to KEGG orthologies)
- Prefix.abundance.txt (abundances of KEGG orthologies)
- Process

Usage: perl FMAP_assembly.pl [options] output.prefix assembly.fasta [input.fastq|input.R1.fastq,input.R2.fastq [...]] > summary.txt
Options: -h display this help message
-A STR prepared assembly prefix
-B input indexed sorted BAM file instead of FASTQ file
-m FILE executable file path of mapping program, "diamond" or "usearch" [diamond]
-p INT number of threads [1]
-e FLOAT maximum e-value to report alignments [10]
-t DIR directory for temporary files [$TMPDIR or /tmp]
-a FLOAT search acceleration for ublast [0.5]
-C STR codon and translation e.g. ATG=M [NCBI genetic code 11 (Bacterial, Archaeal and Plant Plastid)]
-S STR comma-separated start codons [GTG,ATG,CTG,TTG,ATA,ATC,ATT]
-T STR comma-separated termination codons [TAG,TAA,TGA]
-l INT minimum translation length [10]
-c FLOAT minimum coverage [0.8]
-q INT minimum mapping quality [0]
-s STR strand specificity, "f" or "r"
-P STR contig prefix used for abundance estimation
- FMAP_assembly_centrifuge.pl
- Require Centrifuge.
- Input
- FMAP_assembly.region.txt (ORF regions mapping to KEGG orthologies generated by FMAP_assembly)
- De novo assembled sequences in FASTA format
- Centrifuge index filename prefix (minus trailing .X.cf)
- Output: FMAP_assembly.region.taxon.txt (FMAP_assembly.region.txt including a column of NCBI taxonomy IDs (integer))
Usage: perl FMAP_assembly_taxon.pl [options] FMAP_assembly.region.txt assembly.fasta centrifuge.index
Options: -h display this help message
-p INT number of threads [1]
- FMAP_assembly_heatmap.pl
- Require Bio::DB::Taxonomy.
- Input: FMAP_assembly.abundance.txt (abundances generated by FMAP_assembly)
- Output: HTML format of abundance heatmap table
Usage: perl FMAP_assembly_heatmap.pl [options] [name=]FMAP_assembly.abundance.txt [...] > FMAP_assembly_heatmap.html
Options: -h display this help message
-c FILE comparison output file including orthology and filter columns
-f INT HTML font size
-w INT HTML table cell width
- FMAP_assembly_operon.pl
- Input: FMAP_assembly.region.txt (ORF regions mapping to KEGG orthologies generated by FMAP_assembly)
- Output: FMAP_assembly_operon.txt (ODB (v3) known operons consisting of orthologies located together on an assembled contig/scaffold/transcript)
Usage: perl FMAP_assembly_operon.pl [options] FMAP_assembly.region.txt > FMAP_assembly_operon.txt
Options: -h display this help message
-a print single-gene operons as well
- FMAP_download_genome.pl
- Input: NCBI taxonomy IDs (integer)
- Output: FASTA file containing genome sequences
- Require XML::LibXML.
Usage: perl FMAP_download_genome.pl [options] NCBI_TaxID [...] > genome.fasta
Options: -h display this help message
-a assembly instead of genome
- FMAP_download.pl
Usage: perl FMAP_download.pl [options]
Options: -h display this help message
-m FILE executable file path of mapping program, "diamond" or "usearch" [diamond]
-k download prebuilt KEGG files
-x download only KEGG files
- FMAP_mapping.pl
- Input: whole metagenomic (or metatranscriptomic) shotgun sequencing reads in FASTQ or FASTA format
- Output: best-match hits in NCBI BLAST ‑m8 (= NCBI BLAST+ ‑outfmt 6) format
Usage: perl FMAP_mapping.pl [options] input1.fastq|input1.fasta [input2.fastq|input2.fasta [...]] > blastx_hits.txt
Options: -h display this help message
-m FILE executable file path of mapping program, "diamond" or "usearch" [diamond]
-p INT number of threads [1]
-e FLOAT maximum e-value to report alignments [10]
-t DIR directory for temporary files [$TMPDIR or /tmp]
-a FLOAT search acceleration for ublast [0.5]
- FMAP_quantification.pl
- Input: output of "FMAP_mapping.pl"
- Output: abundances (RPKM) of KEGG orthologies
- Output columns: KEGG Orthology ID, orthology definition, abundance (RPKM)
Usage: perl FMAP_quantification.pl [options] blast_hits1.txt [blast_hits2.txt [...]] > abundance.txt
Options: -h display this help message
-c use CPM values instead of RPKM values
-i FLOAT minimum percent identity [80]
-l FILE tab-delimited text file with the first column having protein names and the second column having the sequence lengths
-o FILE tab-delimited text file with the first column having protein names and the second column having the orthology names
-d FILE tab-delimited text file with the first column having orthology names and the second column having the definitions
-w FILE tab-delimited text file with the first column having read names and the second column having the weights
- FMAP_table.pl
- Input: outputs of "FMAP_quantification.pl"
- Output: abundance table
- Output columns: KEGG Orthology ID, orthology definition, abundance of sample1, abundance of sample2, ...
Usage: perl FMAP_table.pl [options] [name1=]abundance1.txt [[name2=]abundance2.txt [...]] > abundance_table.txt
Options: -h display this help message
-c use raw read counts (readCount|count) instead of RPKM values
-d use normalized mean depths (meanDepth/genome) instead of RPKM values
-f use fractions
-n do not print definitions
-r print ORF regions
- FMAP_comparison.pl
- Input: output of "FMAP_table.pl", sample group information
- Output: comparison test statistics for orthologies
- Output columns: KEGG Orthology ID, orthology definition, log2 fold change, p-value, FDR-adjusted p-value, filter (pass or fail)
Usage: perl FMAP_comparison.pl [options] abundance_table.txt control1[,control2[...]] case1[,case2[...]] [...] > orthology_test_stat.txt
Options: -h display this help message
-t STR statistical test for comparing sample groups, "kruskal", "anova", "poisson", "quasipoisson", "metagenomeSeq" [kruskal]
-f FLOAT fold change cutoff [2]
-p FLOAT p-value cutoff [0.05]
-a FLOAT FDR-adjusted p-value cutoff [1]
- FMAP_pathway.pl
- Input: output of "FMAP_comparison.pl"
- Output: pathways enriched in filter-passed orthologies
- Output columns: KEGG Pathway ID, pathway definition, orthology count, coverage, p-value, KEGG Orthology IDs with colors
- KEGG Orthology IDs with colors: input of KEGG Pathway mapping (http://www.kegg.jp/kegg/tool/map_pathway2.html)
Usage: perl FMAP_pathway.pl [options] orthology_test_stat.txt > pathway.txt
Options: -h display this help message
- FMAP_module.pl
- Input: output of "FMAP_comparison.pl"
- Output: modules enriched in filter-passed orthologies
- Output columns: KEGG Module ID, module definition, orthology count, coverage, p-value, KEGG Orthology IDs with colors
- KEGG Orthology IDs with colors: input of KEGG Pathway mapping (http://www.kegg.jp/kegg/tool/map_pathway2.html)
Usage: perl FMAP_module.pl [options] orthology_test_stat.txt > module.txt
Options: -h display this help message
- FMAP_operon.pl
- Input: output of "FMAP_comparison.pl"
- Output: operons consisting of filter-passed orthologies
- Output columns: ODB (v3) known operon IDs, operon definition, log2 fold change, KEGG Orthology IDs, KEGG Pathway IDs
Usage: perl FMAP_operon.pl [options] orthology_test_stat.txt > operon.txt
Options: -h display this help message
-a print single-gene operons as well
- FMAP_plot.pl
- Input: output of "FMAP_pathway.pl", "FMAP_module.pl", or "FMAP_operon.pl"
- Output: PNG format image file of p-value plot
Usage: perl FMAP_plot.pl [options] pathway.txt|module.txt|operon.txt plot.pdf
Options: -h display this help message
-w INT plot width [12]
-h INT plot height [8]
-l FLOAT plot left margin [20]
-p FLOAT p-value cutoff [0.05]
-c FLOAT coverage cutoff [0 for pathway, 1 for module and operons]
-d do not print definition
- FMAP_all.pl
- Input: configuration table file
- Input columns: group (control, ...), sample name, input file of "FMAP_mapping.pl"
- Output: script file including all FMAP commands, all FMAP outputs
Usage: perl FMAP_all.pl [options] input.config [output_prefix]
Options: -h display this help message
-s generate a script, but not execute it
-m FILE mapping: executable file path of mapping program, "diamond" or "usearch" [diamond]
-t INT mapping: number of threads [1]
-c STR comparison: statistical test for comparing sample groups, "kruskal", "anova", "poisson", "quasipoisson", "metagenomeSeq" [kruskal]
-f FLOAT comparison: fold change cutoff [2]
-p FLOAT comparison: p-value cutoff [0.05]
-a FLOAT comparison: FDR-adjusted p-value cutoff [1]
-
Use the prebuilt database (UniRef90 and bacteria/archaea/fungi)
- FMAP_download.pl
- FMAP_mapping.pl
- FMAP_quantification.pl
- FMAP_table.pl
- FMAP_comparison.pl
- FMAP_pathway.pl
- FMAP_module.pl
- FMAP_operon.pl
-
Use a custom database (you can define UniRef and taxonomy.)
- FMAP_database.pl
- FMAP_prepare.pl
- FMAP_mapping.pl
- FMAP_quantification.pl
- FMAP_table.pl
- FMAP_comparison.pl
- FMAP_pathway.pl
- FMAP_module.pl
- FMAP_operon.pl
Kim J, Kim MS, Koh AY, Xie Y, Zhan X. "FMAP: Functional Mapping and Analysis Pipeline for metagenomics and metatranscriptomics studies" BMC Bioinformatics. 2016 Oct 10;17(1):420. PMID: 27724866