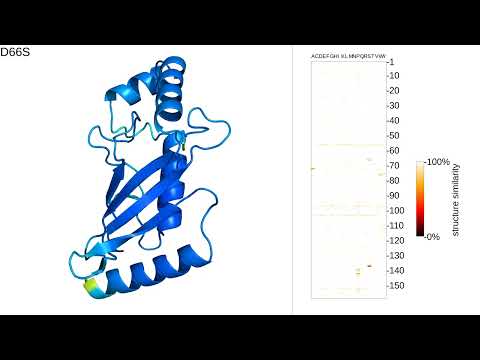
MutAmore visualizes the predicted structural changes of single amino-acid substitutions on a protein sequence as a protein mutation movie (PMM).
Examples rendered in 4K resolution are available on YouTube.
You can try MutAmore on Google Colab in this notebook. The Colab notebook is sufficient for movie rendering of (shorter) protein sequences. If you want to create movies for a larger set of sequences or particularly large proteins, consider installing MutAmore locally as described below.
MutAmore is written in Python and requires the Python packages Pillow, Scipy and Biopython:
pip install Pillow scipy biopython
For movie rendering, you will also need to have PyMOL and ffmpeg installed. These packages are best installed using the package manager of your Linux distribution, e.g. for Debian/Ubuntu systems:
apt-get install pymol
apt-get install ffmpeg
Alternatively, you can use Conda:
conda install -c conda-forge pymol-open-source
conda install -c conda-forge ffmpeg
You will need a structure prediction system for the prediction of mutated structures. We recommend using ESMFold or ColabFold, but any alternative structure predictor can be used (see below).
MutAmore expects one or more amino-acid sequences in FASTA format as input.
python run_movie_rendering.py -i <INPUT_FASTA> -s predictor_scripts/esmfold.sh
The predictor script tells MutAmore how to call the structure prediction system. We provide two scripts for ESMFold and ColabFold.
Before running MutAmore, edit the script predictor_scripts/esmfold.sh and replace ESMFOLD_LOCATION with the path to your ESMFold installation.
Running ColabFold in single-sequence mode works out-of-the-box with the provided script predictor_scripts/colabfold.sh. If you want to run ColabFold with locally generated multiple-sequence alignments, refer to the documentation and include calls to colabfold_search in the predictor script.
Any alternative structure prediction system can be used, as long as it expects FASTA files as inputs and generates PDB files.
Create a new script in predictor_scripts/ and fill in the command-line instruction to run the structure prediction system. Use the placeholders MUTAMORE_INPUT and MUTAMORE_OUTPUT as the input FASTA file and output directory respectively. MutAmore will replace these placeholders at runtime with the appropriate parameters.
Structure prediction systems usually require powerful GPUs and are often run in server environments. Some users might not be able to install graphical packages such as PyMOL and ffmpeg in their server environment. In this case, you can run MutAmore in two seperate steps on different machines. You can run only the structure prediction part of the pipeline by specifying an output directory for the structure predictions:
python run_movie_rendering.py -i <INPUT_FASTA> -s predictor_scripts/esmfold.sh --only-predict -p <PREDICTION_DIRECTORY>
You can then transfer the prediction directory to another machine, e.g. a desktop computer, to run the rendering:
python run_movie_rendering.py -i <INPUT_FASTA> --only-render -p <PREDICTION_DIRECTORY>
MutAmore by default renders movies in the resolution 1280x720. You can set a different resolution with the parameters width and height, e.g. to render in 4K resolution add the following to the MutAmore call:
--width 3840 --height 2160
Instead of creating a movie with all mutants, you can use the parameter top to specify the number of most impactful mutations to be included in the movie, which also reduces rendering time significantly. To e.g. only show the top-50 mutants, use the following parameter:
--top 50
In case you have experimental structures of mutants, you can use them in place of structure predictions. For this, please put the PDB files of mutants in a separate directory with the file name indicating the mutation (e.g. L32K.pdb) and tell MutAmore the directory with the following parameter:
--experimental_dir <STRUCTURE_DIRECTORY>
In this mode, predicted structures will be shown with slightly less opacity to make them visually distinguishable from experimental structures.
MutAmore uses PyMOL to automatically find a proper zoom level. In case you want to override this setting, you can use the parameter -z to specify a zoom offset. Positive numbers cause the camera to zoom out while negative values will zoom further in:
-z 10
MutAmore creates a temporary directory to store intermediate files, which is deleted after finishing. By default, it will create the directory ./tmp/ where you call MutAmore. To specify a different directory, use the parameter -t:
-t <TEMPORARY_DIRECTORY>
MutAmore stores structure predictions in a temporary directory (see above), which is deleted after rendering the movie. In case you want to keep the structure prediction output, you can specify a different directory with the parameter -p:
-p <PREDICTION_DIRECTORY>
By default, MutAmore outputs the finished movies in the directory in which you call MutAmore. To output to a different location, use the parameter -o:
-o <MOVIE_OUTPUT_DIRECTORY>
MutAmore uses predicted confidence scores as obtained from the b-factor column of PDB files for color-coding the 3D visualizations. This way of representing confidence has been established as a standard by AlphaFold2 and as of today been used by all recent 3D structure prediction methods. We expect future methods to follow in line.
The pre-print is available on bioRxiv. Cite with
@article {Weissenow2023,
author = {Weissenow, Konstantin and Rost, Burkhard},
title = {Rendering protein mutation movies with MutAmore},
elocation-id = {2023.09.15.557870},
year = {2023},
doi = {10.1101/2023.09.15.557870},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/10.1101/2023.09.15.557870v1},
eprint = {https://www.biorxiv.org/content/10.1101/2023.09.15.557870v1.full.pdf},
journal = {bioRxiv}
}
or
K. Weissenow and B. Rost, Rendering protein mutation movies with MutAmore, bioRxiv (2023)