The comparator can compare HLA or KIR alleles between cohorts
pip install git+https://github.com/linnil1/star_alleles_comparatorstar_allele_comp hla_result1.csv hla_result2.csv --family hla --save tmp --plot -vThe results will output to screen and save in .txt and .csv format.
The example output is the same as below (see next section).
The input CSV should adhere to the following format:
Columns
id(required): The sample ID.method(optinal): The method. If not specified, filename will be used.allele*(required) Columns starting withallelewill be used to store the allele for each id/sample with corresponding method. The value can be NULL, empty.
For this format, each allele is represented in separate columns:
method,id,allele1,allele2,allele3,allele4
method1,id1,"A*01:02:03:04","A*01:02","B*01:01:01:01"
method1,id2,"A*01:02:03:04","A*01:02","B*01:01:01:01"
method1,id2,"C*03","C*03:03"
method2,id1,"A*01:02:03:04","A*01:02:03","B*01:02:02:01","B*04:01:02"
method2,id2,"A*01:02:03:04","A*01:02:03","B*01:02:02:01","B*04:01:02"
method2,id2,"C*03:03", "C*03:02"
In this format, the alleles column contains a single string with alleles separated by underscores:
method,id,alleles
method1,id3,"KIR2DL1*0010203_KIR2DL1*001_KIR2DS1*0010101"
method1,id4,"KIR2DL1*0010203_KIR2DL1*00102_KIR2DS1*00101"
method1,id3,"KIR2DL1*03105_KIR2DL1*03:03"
method2,id3,"KIR2DL1*001_KIR2DL1*0030203_KIR2DS1*0010208_KIR2DS1*0040102"
method2,id4,"KIR2DL1*0010203_KIR2DL1*0010203_KIR2DS1*0010202_KIR2DS1*0040302"
method2,id4,"KIR2DL1*00303_KIR2DL1*03002"
from star_allele_comp import compare_method, print_all_summary, plot_summary
cohort = {
"method1": { "sample_id1": [ "A*01:02:03:04", "A*01:02", "B*01:01:01:01", "B*03:01"] },
"method2": { "sample_id1": [ "A*01:02:03:04", "A*01:02:03", "B*01:02:02:01", "B*04:01:02"] },
}
ground_truth_method = "method1"
result = compare_method(cohort, ground_truth_method, "hla")print(result)
# Method method2
# Sample sample_id1
# A*01:02:03:04 =4= A*01:02:03:04
# A*01:02 =2= A*01:02:03
# B*01:01:01:01 =1= B*01:02:02:01
# B*03:01 =0= B*04:01:02
# Note:
# Left hand side is the alleles in reference method/cohort
# Right hand side is the allele in another method/cohort# details are in star_allele_comp/summary.py:print_all_summary
df_cohort = result.to_dataframe()
print_all_summary(df_cohort)Accuracy summary
Accuracy num_match num_ref
Resolution 0 1 2 3 4 FP FN 0 1 2 3 4 FP FN 0 1 2 3 4 FP FN
method
method1 1.0 1.00 1.0 1.0 1.0 0.0 0.0 4 4 4 2 2 0 0 4 4 4 2 2 0 0
method2 1.0 0.75 0.5 0.5 0.5 0.0 0.0 4 3 2 1 1 0 0 4 4 4 2 2 0 0
# Note In the accuracy summary table:
# * num_match represents the number of alleles that match the alleles in the ground truth method under the specific `Resolution`.
# * num_ref indicates the number of reference alleles with resolution >= `Resolution`
# * Accuracy is calculated as the ratio of num_match to num_ref.
# * Accuracy in FP is False Discovery Rate (FDR)
# * Accuracy in FN is False Negative Rate (FNR)
Confusion matrix (not the same sample)
Count
match_res -1 0 1 2 3
ref_res
-1 2 0 0 0 0
1 1 1 0 0 0
2 1 0 2 6 0
3 0 0 0 0 1
4 0 0 0 0 1
# Note
# -1 indicates FP or FN
Accuracy summary per resolution per gene
Accuracy num_match num_ref
Resolution 0 1 2 3 4 FP FN 0 1 2 3 4 FP FN 0 1 2 3 4 FP FN
method gene
method1 A 1.0 1.0 1.0 1.0 1.0 0.0 0.0 2 2 2 1 1 0 0 2 2 2 1 1 0 0
B 1.0 1.0 1.0 1.0 1.0 0.0 0.0 2 2 2 1 1 0 0 2 2 2 1 1 0 0figs = plot_summary(df_cohort)
# You can use Dash to show it
from dash import dcc, html, Dash
app = Dash(__name__)
app.layout = html.Div(children=[dcc.Graph(figure=fig) for fig in figs])
app.run(debug=True)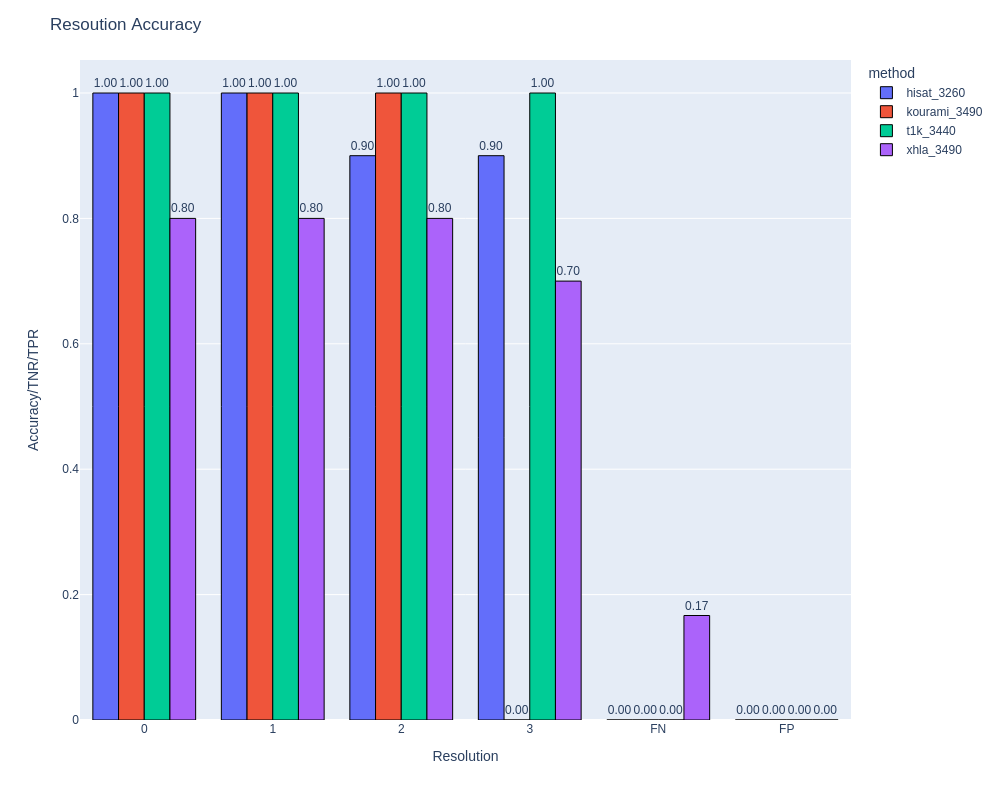
pip install pdoc
pdoc star_allele_comp --docformat google
