This page contains procedures, scripts and analyses developed in:
Genome-scale detection of positive selection in 9 primates predicts human-virus evolutionary conflicts Robin van der Lee, Laurens Wiel, Teunis J.P. van Dam, Martijn A. Huynen
Nucleic Acids Research 2017; https://doi.org/10.1093/nar/gkx704
Centre for Molecular and Biomolecular Informatics - Radboud Institute for Molecular Life Sciences
Radboud university medical center
Nijmegen, The Netherlands
Please consider citing the paper if you found this resource useful.
Note that these scripts were not designed to function as a fully-automated pipeline, but rather as a series of individual steps with extensive manual quality control between them. It will therefore not be straightforward to run all steps smoothly in one go. Feel free to contact me if you run into any issue.
Additional data and material can be found here: Supplementary data and material.
Our scripts depend on various programs and modules to run. Versions mentioned below refer to those used for the analyses described in the paper.
Make sure all of these programs are in your $PATH.
- Perl 5: https://www.perl.org/
- Install various modules using
cpanorcpanmcpanm DBIcpanm DBD::mysql(requires a working MySQL installation, https://www.mysql.com/)
- Download the various helper scripts that are also part of this GitHub repository
functions.pl- Scripts in
Ensembl_API_subroutines
- BioPerl
cpanm Bio::Perl(http://bioperl.org/) - Ensembl API (http://www.ensembl.org/info/docs/api/index.html) - release 78, December 2014 (http://dec2014.archive.ensembl.org/)
GNU Parallel (https://www.gnu.org/software/parallel/)
- PRANK multiple sequence aligner (http://wasabiapp.org/software/prank/) - v.140603
- GUIDANCE (http://guidance.tau.ac.il/) - v1.5
Note that a bug fix is required for GUIDANCE (version 1.5 - 2014, August 7) to work with PRANK, see GUIDANCE_source_code_fix_for_running_PRANK - t_coffee, which includes
TCS(http://www.tcoffee.org/Projects/tcoffee/) - Version_11.00.61eb9e4 - Jalview alignment viewer (http://www.jalview.org/)
PAML software package, which includes codeml (http://abacus.gene.ucl.ac.uk/software/paml.html) - v4.8a
Make sure you check the GNU Parallel logs (perl parse_parallel_logs.pl all) to get an indication that steps finished correctly. Though unfortunately not all programs exit with an error message if they fail, so our scripts also extensively check for whether the various steps ran correctly in other ways.
Please see the Materials and Methods section of the paper for theory and detailed explanations.
Analyses presented in the paper are based on Ensembl release 78, December 2014 (http://dec2014.archive.ensembl.org/).
Obtain one-to-one ortholog clusters for nine primates with high-coverage whole-genome sequences. These scripts can be edited to obtain orthology clusters for (i) a different set of species than the ones we use here, and (ii) different homology relationships than the one-to-one filter we use.
Two methods, same result:
- Fetch orthology information using the Ensembl API:
get_one2one_orthologs__Ensembl_API.pl - Clean the results using:
get_one2one_orthologs__Ensembl_API__process_orthologs.r
- From BioMart, first get orthology information for all species of interest
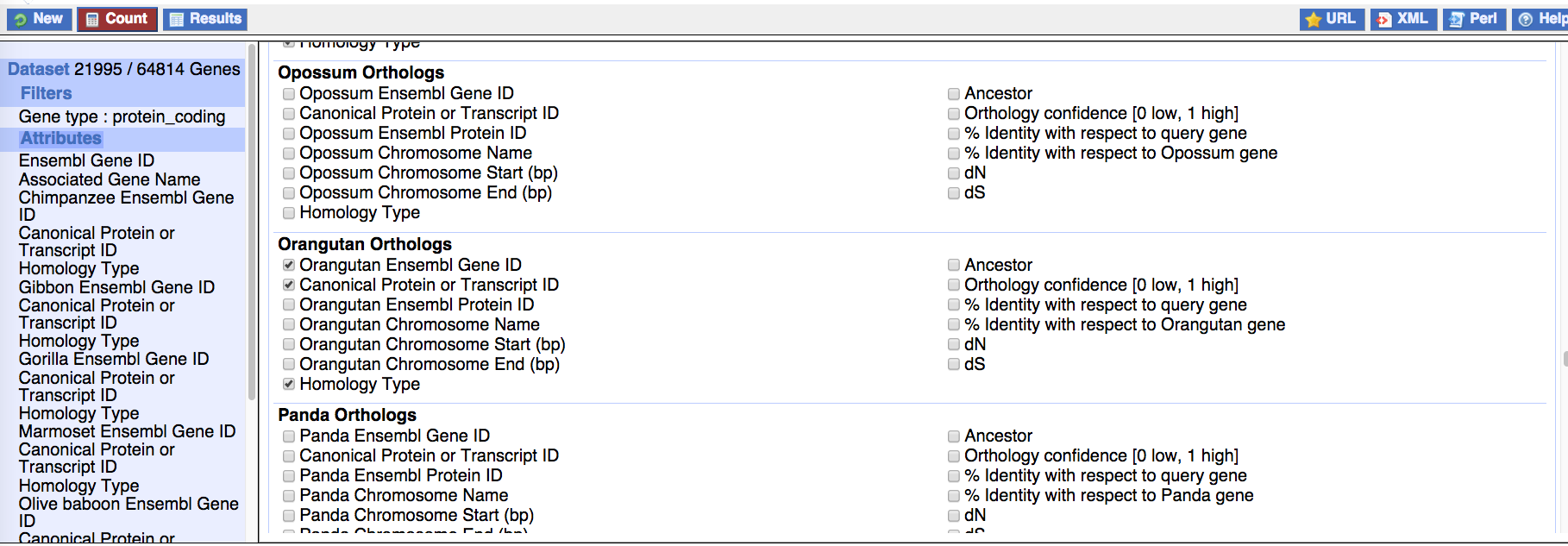
- Combine the acquired ortholog information using
get_one2one_orthologs__combine_biomart_orthology_lists.r


For all one-to-one orthologs, get the coding DNA (cds) and the corresponding protein sequences from the Ensembl Compara gene trees (as these trees are the basis for the orthology calls).
- Run
start_parallel_sequence_fetching.pl, which reads the ortholog cluster information, divides the genes in batches of 1000 ($batchsizecan be changed in the script) and prints the instructions for the next step: get_sequences_for_one2one_orthologs_from_gene_tree_pipeline.pl. This step fetches the sequences. E.g.
STARTING PARALLEL INSTANCE 11
from 10001 to 11000
1000 genes
screen -S i11
perl get_sequences_for_one2one_orthologs_from_gene_tree_pipeline.pl -p -f sequences/parallel_instance_11.txtNote that these steps also fetch the alignments underlying the Compara gene trees, filtered for the species of interest. These are not required for later steps.
check_compatibility_protein_cds_sequences.pl. Tends to only complain at annotated selenocysteine residues.
Produce codon-based nucleotide sequence alignments for all the one-to-one ortholog clusters. Then assess the confidence in the alignments using two independent approaches.
This step finds the fasta files containing cDNA sequences for the species of interest (obtained in Step 2), and for each of them runs the PRANK in codon mode (-codon) to align them. Jobs are executed and monitored in parallel using GNU Parallel (set number of cores with --max-procs).
find sequences/ -type f -name "*__cds.fa" | parallel --max-procs 4 --joblog parallel_prank-codon.log --eta 'prank +F -codon -d={} -o={.}.prank-codon.aln.fa -quiet > /dev/null'This effectively executes the following commands:
find sequences/ -type f -name "*__cds.fa" | parallel 'echo prank +F -codon -d={} -o={.}.prank-codon.aln.fa -quiet'
...
prank +F -codon -d=sequences//cds/ENSG00000274211__cds.fa -o=sequences//cds/ENSG00000274211__cds.prank-codon.aln.fa -quiet
prank +F -codon -d=sequences//cds/ENSG00000274523__cds.fa -o=sequences//cds/ENSG00000274523__cds.prank-codon.aln.fa -quiet
prank +F -codon -d=sequences//cds/ENSG00000019549__cds.fa -o=sequences//cds/ENSG00000019549__cds.prank-codon.aln.fa -quiet
...NOTE: this step takes a lot of computation time.
- Run GUIDANCE to assess the sensitivity of the alignment to perturbations of the guide tree.
Note that this requires a bug fix in GUIDANCE (version 1.5 - 2014, August 7), see GUIDANCE_source_code_fix_for_running_PRANK
find sequences/ -type f -name "*__cds.fa" | parallel --max-procs 4 --nice 10 --joblog parallel_guidance-prank-codon.log --eta 'mkdir -p sequences/guidance-prank-codon/{/.}; guidance.pl --program GUIDANCE --seqFile {} --seqType nuc --msaProgram PRANK --MSA_Param "\+F \-codon" --outDir sequences/guidance-prank-codon/{/.} &> sequences/guidance-prank-codon/{/.}/parallel_guidance-prank-codon.output'mask_msa_based_on_guidance_results.pl. Analyze and parse the GUIDANCE results. Low confidence scores led us to remove entire alignments from our analysis or mask unreliable individual columns and codons.
Run T-Coffee TCS to assess alignment stability by independently re-aligning all possible pairs of sequences. Note that we ran TCS on translated PRANK codon alignments.
- Translate the PRANK cDNA alignments to protein alignments. Note that we use the PRANK alignments generated through GUIDANCE (Step 3b) to ensure we are masking the same alignments with both GUIDANCE and TCS!
mkdir -p sequences/tcs-prank-codon
find sequences/prank-codon-masked/ -type f -name "*__cds.prank-codon.aln.fa" | parallel --max-procs 4 --nice 10 --joblog parallel_translate-prank-codon-alignments.log --eta 't_coffee -other_pg seq_reformat -in {} -action +translate -output fasta_aln > sequences/tcs-prank-codon/{/.}.translated.fa'- Run TCS on the translated PRANK alignments:
cd sequences/tcs-prank-codon
find . -type f -name "*prank-codon.aln.translated.fa" | parallel --max-procs 4 --nice 10 --joblog ../../parallel_tcs-t-coffee.log --eta 't_coffee -infile {} -evaluate -method proba_pair -output score_ascii, score_html -quiet > /dev/null'
cd ../../mask_msa_based_on_tcs_results.pl. Analyze and parse the TCS results. Low confidence scores led us to remove entire alignments from our analysis or mask unreliable individual columns and codons. Note that we mask the original PRANK codon-based cDNA alignments based on the TCS results on the translated alignment!
- Sort sequences within alignment fasta files by species using
sort_sequences_by_taxon.pl, so that all alignment files have the same sequence ordering.
cd sequences/prank-codon-masked/
find . -type f -name "*prank-codon-guidance-tcs-masked.aln.fa" | parallel --max-procs 4 --joblog ../../parallel_sort_alignments.log --eta --colsep '__cds' 'perl ../../sort_sequences_by_taxon.pl {1}__cds{2} {1}__cds.prank-codon-guidance-tcs-masked-species-sorted.aln.fa'
cd ../../- Translate masked cDNA alignments to facilitate quality control and visualization:
find sequences/prank-codon-masked/ -type f -name "*__cds.prank-codon-guidance-tcs-masked-species-sorted.aln.fa" | parallel --max-procs 4 --nice 10 --joblog parallel_translate-prank-codon-guidance-tcs-masked-species-sorted-alignments.log --eta 't_coffee -other_pg seq_reformat -in {} -action +translate -output fasta_aln > sequences/prank-codon-masked/{/.}.translated.fa'Perform maximum likelihood (ML) dN/dS analysis to infer positive selection of genes and codons, using codeml from the PAML software package.
Construct a single phylogenetic tree with branch lengths for use in the codeml analyses of all individual one-to-one ortholog cluster alignments (Step 4b, below).
-
perl concatenate_alignments.pl. Concatenate all 11,096 masked alignments from Step 3 (i.e. the GUIDANCE- and TCS-masked codon-based cDNA alignments) into one large alignment. First make sure individual alignment files are all sorted in the same way (done in Step 3d). -
Sort sequences within the concatenated alignment again by species:
perl sort_sequences_by_taxon.pl sequences/concatenated_alignment__9primates__cds.prank-codon-guidance-tcs-masked.aln.fa sequences/concatenated_alignment__9primates__cds.prank-codon-guidance-tcs-masked-species-sorted.aln.fa- Convert concatenated alignment from FASTA to a PHYLIP format that is compatible with PAML codeml. This script checks that (i) sequence names do not contain characters that cannot be handled by codeml, (ii) sequences do not contain stop codons or non-canonical nucleotides, (iii) undetermined and masked codons [nN] are converted to the codeml ambiguity character
?.
perl convert_fasta_to_codeml_phylip.pl sequences/concatenated_alignment__9primates__cds.prank-codon-guidance-tcs-masked-species-sorted.aln.fa- Run the codeml M0 model on the concatenated alignment. This fits a single dN/dS to all sites (
NSsites = 0, model = 0, method = 1, fix_blength = 0). We provided codeml with the well-supported topology of the primate phylogeny: Ensembl78__9primates__with_taxon_id__unrooted.tre. See the.ctlfiles for exact configurations: Configuration_files_for_PAML_codeml.
- Calculate reference tree under the F3X4 codon frequency parameter:
mkdir codeml_M0_F3X4
cd codeml_M0_F3X4/
cp ../Supplementary_data_and_material/Phylogenetic_Trees/Ensembl78__9primates__with_taxon_id__unrooted.tre .
cp ../Supplementary_data_and_material/Configuration_files_for_PAML_codeml/codeml_M0_F3X4_tree.ctl .
codeml codeml_M0_F3X4_tree.ctl > codeml_M0_F3X4_tree.screen_output
cd ..- Calculate reference tree under the F61 codon frequency parameter:
mkdir codeml_M0_F61
cd codeml_M0_F61/
cp ../Supplementary_data_and_material/Phylogenetic_Trees/Ensembl78__9primates__with_taxon_id__unrooted.tre .
cp ../Supplementary_data_and_material/Configuration_files_for_PAML_codeml/codeml_M0_F61_tree.ctl .
codeml codeml_M0_F61_tree.ctl > codeml_M0_F61_tree.screen_output
cd ..- Store the phylogenetic trees outputted by codeml. Ours are codeml_M0_tree__unrooted_tree__F3X4.tre and codeml_M0_tree__unrooted_tree__F61.tre.
- Convert individual codon-based cDNA alignments from FASTA to a PHYLIP format that is compatible with PAML codeml. This script (i) checks that sequence names do not contain characters that cannot be handled by codeml, (ii) removes gene identifiers from the sequence IDs to make all
.phyfiles compatible with the species names in the phylogenetic tree supplied to codeml, (iii) checks that sequences do not contain stop codons or non-canonical nucleotides, (iv) converts undetermined and masked codons [nN] to the codeml ambiguity character?.
find sequences/prank-codon-masked/ -type f -name "*__cds.prank-codon-guidance-tcs-masked-species-sorted.aln.fa" | parallel --max-procs 4 --nice 10 --joblog parallel_convert-to-phylip-prank-codon-guidance-tcs-masked-species-sorted.log --eta 'perl convert_fasta_to_codeml_phylip.pl {}'- Run codeml. NOTE: this step takes a lot of computation time.
These commands (i) prepare the directory structure, (ii) copy the template codeml .ctl file and the reference phylogenetic tree to the proper directories, (iii) customize the .ctl files for running the analysis on each of the 11,096 alignments, and eventually (iv) run the codeml program (from within start_codeml_for_single_alignment.pl).
The following code shows how to run the M7vM8_F61 parameter combination. For the paper, we used four combinations of the following codeml .ctl file parameters: NSsites = 1 2 or NSsites = 7 8; CodonFreq = 2 or CodonFreq = 3.
To run the other three parameter combinations:
- In the code below, replace
M7vM8_F61byM1avM2a_F61 - In the code below, replace
M7vM8_F61byM7vM8_F3X4andcodeml_M0_tree__unrooted_tree__F61.trebycodeml_M0_tree__unrooted_tree__F3X4.tre - In the code below, replace
M7vM8_F61byM1avM2a_F3X4andcodeml_M0_tree__unrooted_tree__F61.trebycodeml_M0_tree__unrooted_tree__F3X4.tre
mkdir codeml_M7vM8_F61
cp start_codeml_for_single_alignment.pl codeml_M7vM8_F61/
cd codeml_M7vM8_F61/
cp ../Supplementary_data_and_material/Configuration_files_for_PAML_codeml/codeml_M7vM8_F61__large-scale-analysis__template.ctl .
cp ../Supplementary_data_and_material/Phylogenetic_Trees/codeml_M0_tree__unrooted_tree__F61.tre .
find ../sequences/prank-codon-masked/ -type f -name "*__cds.prank-codon-guidance-tcs-masked-species-sorted.aln.phy" | parallel --max-procs 4 --nice 10 --joblog parallel_codeml__M7vM8_F61.log --eta 'perl start_codeml_for_single_alignment.pl {} codeml_M7vM8_F61__large-scale-analysis__template.ctl'
cd ..process_single_run_codeml_results.plprocesses the results for the individual analyses: (i) extensive checks to see if codeml ran correctly, (ii) copies relevant result files, (iii) parses the relevant results from the various mysteriouscodemloutput files, (iv) combines everything into results tables.
cd codeml_M7vM8_F61/
mkdir codeml_results_parsed
find codeml_results/ -type d | grep ENSG | parallel --max-procs 4 --joblog parallel_parse_codeml_results__M7vM8_F61.log 'perl ../process_single_run_codeml_results.pl {}' &> parallel_parse_codeml_results__M7vM8_F61.output- The results of the individual analyses are then collected into two big tables, one for the alignment level results and one for the residue level results:
find codeml_results_parsed/ -name "*residues_codeml_results" | sort | xargs cat > M7vM8_F61.residues_codeml_results
find codeml_results_parsed/ -name "*alignment_codeml_results" | sort | xargs cat > M7vM8_F61.alignment_codeml_results
cd ..Again, to run the other three parameter combinations:
- In this Step 4c code, replace
M7vM8_F61byM1avM2a_F61 - In this Step 4c code, replace
M7vM8_F61byM7vM8_F3X4 - In this Step 4c code, replace
M7vM8_F61byM1avM2a_F3X4
Combines the results of the different parameter combination runs to select alignments and residues that fulfill our stringent confidence criteria and are thus inferred to have evolved under positive selection. These criteria are the following (refer to the paper for more details):
- The likelihood ratio test (LRT) indicates that the selection model provides a significantly better fit to the data than does the neutral model (P < 0.05, after Benjamini Hochberg correction for testing 11,096 genes). We included apparent Positively Selected Genes (aPSG) if they met the LRT significance criteria under all four tested ML parameter combinations.
- We included apparent Positively Selected Residues (aPSR) if their codons were assigned high Bayesian (BEB) posteriors under all four ML parameter combinations (Pposterior (ω > 1) > 0.99).
- Collect the base result files from the previous step.
mkdir codeml_results_combined
mkdir codeml_results_combined/codeml_results_base_files
find . -name "M*_codeml_results" | parallel 'cp {} codeml_results_combined/codeml_results_base_files'analyze_and_combine_codeml_results.r. Combines base result files, performs P value and multiple testing calculations, and selects the final set of alignments and residues fulfilling the criteria.
To assess the reliability of our procedure we performed systematic manual inspection of all aPSR and aPSG. Details are described in the paper. Alignment visualization is described in the Supplementary data and material: Visualization_and_quality_control_of_alignments_and_positive_selection_profiles