The package locuscomparer is an R package for visualization of GWAS-eQTL colocalization events.
Use the following commands to install LocusCompareR:
install.packages("devtools")
library(devtools)
install_github("boxiangliu/locuscomparer")
library(locuscomparer)
In addition, you will need to install plink. You will also need to download 1000 Genomes genotypes for LD calculation. You can run this file to download the 1000 Genomes genotypes. Note that they are very large.
The input format to locuscompare is a two-column tab-delimited text file. Here is an example file:
rsid pval
rs62156064 0.564395
rs7562234 0.399642
rs11677377 0.34308
rs35076156 0.625237
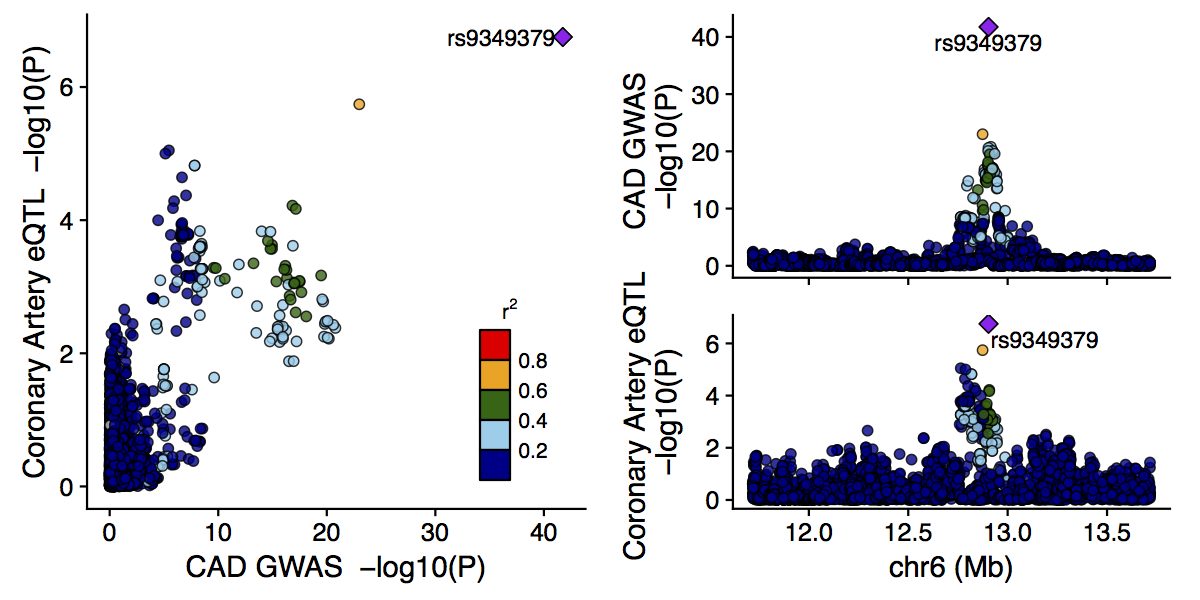
To illustrate the use of locuscompare, we use the Nikpay et al. (2015) and the coronary artery eQTL dataset from GTEx. First download these example GWAS and eQTL datasets.
Then run the following commands:
library(locuscomparer)
gwas = read.table('PHACTR1_gwas.tsv')
eqtl = read.table('Coronary_Artery_PHACTR1_eqtl.tsv')
main(in_fn1 = gwas, in_fn2 = eqtl, title = 'GWAS', title2 = 'eQTL', vcf_fn = 'ALL.chr6.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz')
The file ALL.chr6.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz can be downloaded from 1000 Genomes through FTP.
You should see a figure like this.