![]()

Efficient implementations of Needleman-Wunsch and other sequence alignment algorithms written in Rust with Python bindings via PyO3.
sequence_align is distributed via PyPi for Python 3.7+, making installation as simple as the following --
no special setup required for cross-platform compatibility, Rust installation, etc.!
pip install sequence_alignAlternatively, if one wishes to develop for sequence_align, first ensure that both
Python and Rust
are installed on your system. Then, install Maturin and run
maturin develop (optionally with the -r flag to compile a release build, instead of an unoptimized debug build)
from the root of your cloned repo to build and install sequence_align in your active Python environment.
Pairwise sequence algorithms are available in sequence_align.pairwise.
Currently, two algorithms are implemented: the Needleman-Wunsch algorithm
and Hirschberg’s algorithm. Needleman-Wunsch is
commonly used for global sequence alignment, but suffers from the fact that it uses O(M*N) space,
where M and N are the lengths of the two sequences being aligned. Hirschberg’s algorithm modifies Needleman-Wunsch
to have the same time complexity (O(M*N)), but only use O(min{M, N}) space, making it an appealing option
for memory-limited applications or extremely large sequences.
Using these algorithms is straightforward:
from sequence_align.pairwise import hirschberg, needleman_wunsch
# See https://en.wikipedia.org/wiki/Needleman%E2%80%93Wunsch_algorithm#/media/File:Needleman-Wunsch_pairwise_sequence_alignment.png
# Use Needleman-Wunsch default scores (match=1, mismatch=-1, indel=-1)
seq_a = ["G", "A", "T", "T", "A", "C", "A"]
seq_b = ["G", "C", "A", "T", "G", "C", "G"]
aligned_seq_a, aligned_seq_b = needleman_wunsch(
seq_a,
seq_b,
match_score=1.0,
mismatch_score=-1.0,
indel_score=-1.0,
gap="_",
)
# Expects ["G", "_", "A", "T", "T", "A", "C", "A"]
print(aligned_seq_a)
# Expects ["G", "C", "A", "_", "T", "G", "C", "G"]
print(aligned_seq_b)
# See https://en.wikipedia.org/wiki/Hirschberg%27s_algorithm#Example
seq_a = ["A", "G", "T", "A", "C", "G", "C", "A"]
seq_b = ["T", "A", "T", "G", "C"]
aligned_seq_a, aligned_seq_b = hirschberg(
seq_a,
seq_b,
match_score=2.0,
mismatch_score=-1.0,
indel_score=-2.0,
gap="_",
)
# Expects ["A", "G", "T", "A", "C", "G", "C", "A"]
print(aligned_seq_a)
# Expects ["_", "_", "T", "A", "T", "G", "C", "_"]
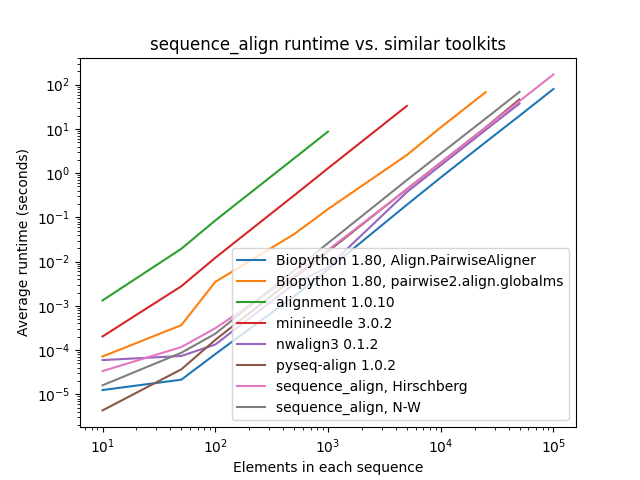
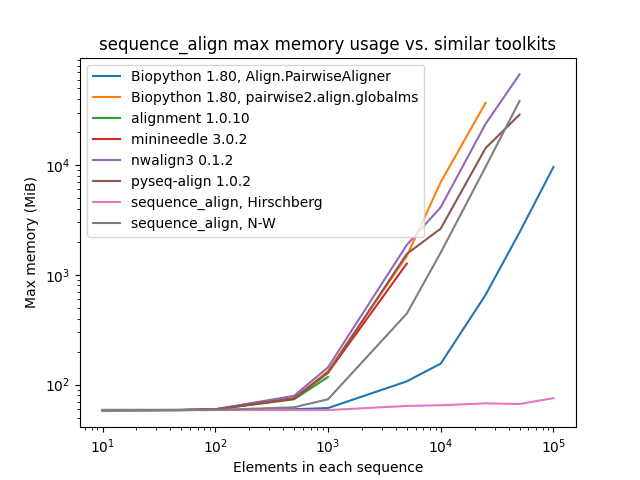
print(aligned_seq_b)All tests below were conducted sequentially on a AWS R5.4 instance with 16 cores and 128 GB of memory. The pair of sequences for alignment consist of a character sequence of randomly selected A/C/G/T nucleotide bases along with another that is identical, except with 10% of the characters randomly perturbed by deletion, insertion of another randomly-selected character after the entry, or replacement with a different randomly-selected character.
As one can see, while sequence_align is comparable to some other toolkits in terms of speed, its memory performance
is best-in-class, even when compared to toolkits using the same algorithm, such as Needleman-Wunsch being used in
pyseq-align.
(Please note that some lines terminate early, as some toolkits took prohibitively long and/or ran out of memory at higher scales.)
Licensed under the Apache 2.0 License. Unless required by applicable law or agreed to in writing, software distributed under the License is distributed on an "AS IS" BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied. See the License for the specific language governing permissions and limitations under the License.
Copyright 2023-present Kensho Technologies, LLC. The present date is determined by the timestamp of the most recent commit in the repository.