PBxplore is a suite of tools dedicated to Protein Block (PB) analysis. Protein Blocks are structural prototypes defined by de Brevern et al. The 3-dimensional local structure of a protein backbone can be modelized as an 1-dimensional sequence of PBs. In principle, any conformation of any amino acid could be represented by one of the sixteen available Protein Blocks (see Figure 1).
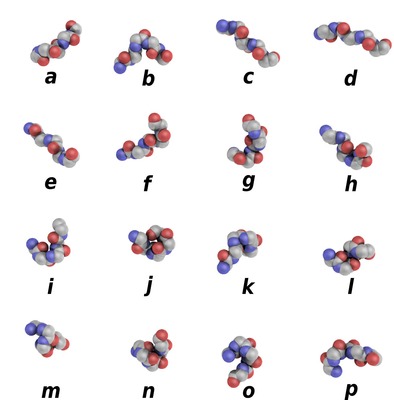
Figure 1. Schematic representation of the sixteen protein blocks, labeled from a to p (Creative commons 4.0 CC-BY).
PBxplore provides both a Python library and command-line tools. Basically, PBxplore can:
- assign PBs from a single PDB, many PDBs or a molecular dynamics simulation trajectory.
- use analysis tools to perform statistical analysis on PBs.
- use analysis tools to study protein flexibility and deformability.
PBxplore requires:
- Python 3.x (>= 3.6)
- Python modules: NumPy, Matplotlib, MDAnalysis (version >= 0.11).
Optionally, PBxplore can use:
- WebLogo 3 to create logo from PB sequences.
Once dependencies installed, the most straightforward way is to use pip:
$ pip install pbxplorePBxplore can also be installed for the current user only:
$ pip install --user pbxploreAll documentation are hosted by Read The Docs and can be found here.
If you use PBxplore, please cite this tool as:
The published version (1.3.8) is archived in Zenodo
and Software Heritage
PBxplore is a research software and has been developped by:
- Pierre Poulain, "Mitochondria, Metals and Oxidative Stress" group, Institut Jacques Monod, UMR 7592, Univ. Paris, CNRS, France.
- Jonathan Barnoud, University of Groningen, Groningen, The Netherlands.
- Hubert Santuz, Laboratoire de Biochimie Théorique, CNRS UPR 9080, Institut de Biologie Physico-Chimique, Paris, France.
- Alexandre G. de Brevern & Gabriel Cretin, DSIMB, INSERM, UMR_S 1134, INTS, Univ Paris, Paris, France.
If you want to report a bug, request a feature, use the GitHub issue system.
PBxplore is licensed under The MIT License.