
Many tools have been developed for the discovery and annotation of transposable elements (TEs). However, the high-quality TE consensus library construction still requires manual curation of TEs, which is time-consuming and needs experts with an in-depth understanding of TE biology.
TEtrimmer is a powerful software designed to automate the manual curation of TEs. The input can be a TE library from de novo TE discovery tools, such as EDTA2 and RepeatModeler2, or a TE library from closely related species. For each input consensus sequence, TEtrimmer automatically performs BLAST, sequence extraction, extension, multiple sequence alignment (MSA), MSA clustering, MSA cleaning, TE boundary definition, and TE classification. TEtrimmer also provides a graphical user interface (GUI) to inspect and improve predicted TEs, which can assist achieving manual curation-level TE consensus libraries easily.

For detailed instructions, including installation steps, usage options, example outputs, and more, please refer to TEtrimmerv1.4.0Manual.pdf
TEtrimmer can be installed by 1. Conda, 2. Singularity, or 3. Docker.
1. Conda (Many thanks to HangXue)
You have to install miniconda on your computer in advance.
We highly recommend installation with mamba, as it is much faster.
# Create new conda environment
conda create --name TEtrimmer
# Install mamba
conda install -c conda-forge mamba
# Activate new environment
conda activate TEtrimmer
# Install TEtrimmer
mamba install bioconda::tetrimmer
# Display options of TETrimmer
TEtrimmer --help
# If you encounter "ClobberError" or "ClobberWarning", don't worry! wait until it is finished!
# The Error or Warning could be like this:
ClobberError: This transaction has incompatible packages due to a shared path.
packages: bioconda/osx-64::blast-2.5.0-boost1.64_2, bioconda/osx-64::rmblast-2.14.1-hd94f91d_0
path: 'bin/blastx'
or See required dependencies TEtrimmer_dependencies.
or conda installation via .yml
# Clone the github repository for TEtrimmer.
git clone https://github.com/qjiangzhao/TEtrimmer.git
# Install mamba
conda install -c conda-forge mamba
# Install TEtrimmer by the "yml" file
mamba env create -f <path to/TEtrimmer_env_for_linux.yml>
Here is the provided TEtrimmer_env_for_linux.yml
# Download and generate TEtrimmer "sif" file
singularity pull docker://quay.io/biocontainers/tetrimmer:1.4.0--hdfd78af_0
# Run TEtrimmer based on sif file
# If <your_path_to_store_PFAM_database> doesn't contain PFAM database
# TEtrimmer can automatically download PFAM to <your_path_to_store_PFAM_database>
singularity exec --writable-tmpfs \
--bind <your_path_contain_genome_file>:/genome \
--bind <your_path_contain_input_TE_library_file>:/input \
--bind <your_output_path>:/output \
--bind <your_path_to_store_PFAM_database>:/pfam \
<your_path_contain_sif_file>/tetrimmer_1.4.0--hdfd78af_0.sif \
TEtrimmer \
-i /input/<TE_library_name.fasta> \
-g /genome/<genome_file_name.fasta> \
-o /output \
--pfam_dir /pfam \
-t 20 --classify_all
# Download TEtrimmer docker image
docker pull quay.io/biocontainers/tetrimmer:1.4.0--hdfd78af_0
docker run -it --name TEtrimmer -v <bind_your_path>:/data quay.io/biocontainers/tetrimmer:1.4.0--hdfd78af_0
# Then you can run TEtrimmer inside TEtrimmer container
# Please note: Run TEtrimmer via Docker is relatively slower than Conda and Singularity.
System: Linux, macOS, Windows WSL
RAM:
- For HPC Linux user, enough RAM needs to be assigned. We highly recommend running TEtrimmer on HPC with at least 40 threads and assigning at least 5 GB RAM to each thread.
| Threads | RAM |
|---|---|
| 40 | 200 GB |
| 100 | 600 GB |
- Windows and macOS PC users can use Virtual Memory. Simply assign 20 threads to push the CPU to its limits. We did tests on a Macbook Pro (2020 M1 chip, 16 GB RAM) and compared with HPC, you can find the running time here:
| Query sequence number | Platform | Threads | RAM | Run time |
|---|---|---|---|---|
| 1700 | Macbook Pro M1 | 20 | 16 GB + Virtual Memory | 50 hours |
| 1700 | HPC | 40 | 150 GB | 5 hours |
- We have not tested it on the WLS of Windows, but it should be feasible to run TEtrimmer on it as well given sufficient resources.
# To see all options
TEtrimmer --help
or
# To see all options
python <path to TEtrimmer>/TEtrimmer.py --help
- Download the test files test_input.fa and test_genome.fasta.
TEtrimmer --input_file <path to test_input.fa> \
--genome_file <path to test_genome.fasta> \
--output_dir <output directory> \
--num_threads 20
--classify_all
- Genome file: The genome sequence in FASTA format (.fa or .fasta).
- TE consensus library: TEtrimmer uses the TE consensus library from de novo TE annotation tools, like
RepeatModelerorEDTA, as input. For this reason, you have to runRepeatModeleror other TE annotation software first.
# TEtrimmer package already includes RepeatModeler. Below is an exmpale command of running RepeatModeler.
# Build genome database index files
BuildDatabase -name <genome_file_database_name> <genome_file.fa>
# Run RepeatModeler
RepeatModeler -database <genome_file_database_name> \
-threads 20 \
-LTRStruct
# Then you will get the TE_consensus_library.fa file
Example:
TEtrimmer --input_file <TE_consensus_library.fa> \
--genome_file <genome_file.fa> \
--output_dir <output_directory> \
--num_threads 20 \
--classify_all
If you want to continue the analysis based on previous unfinished results in the same directory::
TEtrimmer --input_file <TE_consensus_library.fa> \
--genome_file <genome_file.fa> \
--output_dir <directory_contains_previous_unfinished_results> \
--num_threads 20 \
--classify_all \
--continue_analysis
If you want to combine files from different sources for the input file, we recommend removing duplicate sequences before processing. This step can potentially save overall run time in the input file (TEtrimmer only accepts single file input, you have to combine files in advance):
TEtrimmer --input_file <TE_consensus_library.fa> \
--genome_file <genome_file.fa> \
--output_dir <output_directory> \
--num_threads 20 \
--classify_all
--dedup
More options are available:
--classify_unknown Use RepeatClassifier to classify the consensus sequence if the input
sequence is not classified or is unknown or the processed sequence
length by TEtrimmer is 2000 bp longer or shorter than the query
sequence.
--cons_thr FLOAT The minimum level of agreement required at a given position in the
alignment for a consensus character to be called. Default: 0.8
--mini_orf INTEGER Define the minimum ORF length to be predicted by TEtrimmer. Default:
200
--max_msa_lines INTEGER Set the maximum number of sequences to be included in a multiple
sequence alignment. Default: 100
-ga, --genome_anno Perform genome TE annotation using RepeatMasker with the TEtrimmer
curated TE libraries.
- 📁Classification - This folder is used for TE classifications.
- 📁Multiple_sequence_alignment - All raw files will be stored in this folder if < --debug > is enabled.
- 📄error_file.txt - Error file to store all error messages, only visible if errors were found.
- 📁Single_fasta_files - All sequences in the input file will be separated into single FASTA files and stored here.
- 📁TEtrimmer_for_proof_curation - This folder contains files used for manual inspection of TEtrimmer annotations.
- 📁Annotation_perfect - Four files are associated with each sequence as showed below.
- 📄TE_name.raw.fa - Multiple sequence alignment file before TE boundary definition.
- 📄TE_name.fa - Multiple sequence alignment file after TE boundary definition, which is used to generate the consensus sequence.
- 📄TE_name.pdf - Plot file used to evaluate output.
- 📄TE_name.cluster.fa - Multiple sequence alignment file before clustering.
- 📁Annotation_good
- 📁Annotation_check_recommended
- 📁Annotation_check_required
- 📁Clustered_proof_curation - This folder contains all the output files from folder "Annotation_perfect", "Annotation_good", "Annotation_check_recommended", and "Annotation_check_required". The difference is TEtrimmer group similar output TEs into one "Cluster", which can make it easier to compare similar outputs.
- 📁TE_low_copy - This folder contains low copy TEs.
- 📁TE_skipped - Contains TE_Aid plots for all skipped TEs.
- 📁TEtrimmer_proof_anno_GUI - The folder contains graphical user interface tools for manual proof curation.
- 📄annoGUI.py - Use <python ./annoGUI.py -g <genome.fa>> to start manual proof curation GUI.
- 📁Annotation_perfect - Four files are associated with each sequence as showed below.
- 📁HMM - This folder is used to store Hidden Markov Model file. Only visible when < --hmm > is enabled.
- 📄Sequence_name_mapping.txt - This file connects the input sequence names with the modified names from TEtrimmer.
- 📄summary.txt - Summary file.
- 📄TEtrimmer_consensus.fasta - TE consensus library file before de-duplication.
- 📄TEtrimmer_consensus_merged.fasta - TE consensus library file after de-duplication.
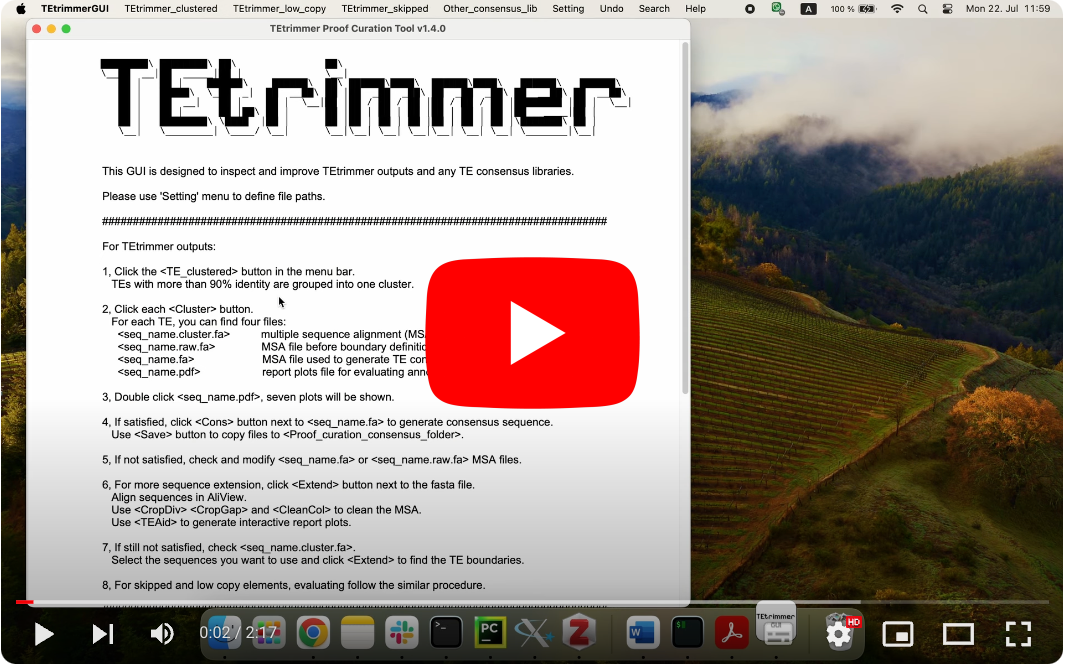
You can use the TEtrimmerGUI tool to inspect and improve TEtrimmer generated TE consensus library. This step is optional! TEtrimmer output can be used for genome-wide TE annotation directly. But if you want to get a traditional manual-curation level TE consensus library, you have to perform this step.
TEtrimmerGUI executable file can be downloaded:
Linux x64: TEtrimmerGUI_Linux
Windows x64: TEtrimmerGUI_Windows.exe
macOS ARM: TEtrimmerGUI_macOS (x86_64 has not been tested.)
For macOS, please run the following command first after decompression:
xattr -d com.apple.quarantine <your_path>/TEtrimmerGUI.app
# We are trying to notarize the app.
Installation is not required, unpack and double-click the executable file to start TEtrimmerGUI. It may take around 30s for initialization after double-clicking "TEtrimmerGUI", please be patient for the first time. You can put the executable file on your desktop or launchpad to make it easier to start it.
If you don't want to download executable files, you can clone the source code and run:
# Use --help to see all options
python <your_path>/tetrimmer/TEtrimmer_proof_anno_GUI/annoGUI.py --help
# To start the manual inspection GUI tool
# Open your Linux, macOS, or Windows terminal and type
python <your_path>/tetrimmer/TEtrimmer_proof_anno_GUI/annoGUI.py
# Note: You have to make BLAST available.
You can easily check and improve TEtrimmer outputs and get manual curation level TE consensus library.
TEtrimmer GUI tool can extend MSA, generate interactive plots, and clean MSA. A demo video is provided.

For each TEtrimmer output TE consensus sequence. You will get a report plot file like this:

Many thanks to all the people who contributed to the TEtrimmer development.
Options:
-i, --input_file TEXT Path to TE consensus file (FASTA format). Use the output from
RepeatModeler, EDTA, REPET, et al. [required]
-g, --genome_file TEXT Path to genome FASTA file (FASTA format). [required]
-o, --output_dir TEXT Path to output directory. Default: current working directory.
-s, --preset [conserved|divergent]
Choose one preset config (conserved or divergent).
-t, --num_threads INTEGER Thread number used for TEtrimmer. Default: 10
--classify_unknown Use RepeatClassifier to classify the consensus sequence if the input
sequence is not classified or is unknown or the processed sequence
length by TEtrimmer is 2000 bp longer or shorter than the query
sequence.
--classify_all Use RepeatClassifier to classify every consensus sequence. WARNING:
This may take a long time.
-ca, --continue_analysis Continue from previous unfinished TEtrimmer run and would use the
same output directory.
--dedup Remove duplicate sequences in input file.
-ga, --genome_anno Perform genome TE annotation using RepeatMasker with the TEtrimmer
curated TE libraries.
--hmm Generate HMM files for each processed consensus sequence.
--debug debug mode. This will keep all raw files. WARNING: Many files will be
generated.
-pd, --pfam_dir TEXT Pfam database directory. TE Trimmer will download the database
automatically. Only turn on this option if you want to use a local
PFAM database or the automatic download fails.
--cons_thr FLOAT The minimum level of agreement required at a given position in the
alignment for a consensus character to be called. Default: 0.8
--mini_orf INTEGER Define the minimum ORF length to be predicted by TEtrimmer. Default:
200
--max_msa_lines INTEGER Set the maximum number of sequences to be included in a multiple
sequence alignment. Default: 100
--top_msa_lines INTEGER If the sequence number of multiple sequence alignment (MSA) is
greater than <max_msa_lines>, TEtrimmer will first sort sequences by
length and choose <top_msa_lines> number of sequences. Then,
TEtrimmer will randomly select sequences from all remaining BLAST
hits until <max_msa_lines>sequences are found for the multiple
sequence alignment. Default: 100
--min_seq_num INTEGER The minimum blast hit number required for the input sequence. We do
not recommend decreasing this number. Default: 10
--min_blast_len INTEGER The minimum sequence length for blast hits to be included for further
analysis. Default: 150
--max_cluster_num INTEGER The maximum number of clusters assigned in each multiple sequence
alignment. Each multiple sequence alignment can be grouped into
different clusters based on alignment patterns WARNING: using a
larger number will potentially result in more accurate consensus
results but will significantly increase the running time. We do not
recommend increasing this value to over 5. Default: 2
--ext_thr FLOAT The threshold to call “N” at a position. For example, if the most
conserved nucleotide in a MSA columnhas proportion smaller than
<ext_thr>, a “N” will be called at this position. Used with
<ext_check_win>. The lower the value of <ext_thr>, the more likely to
get longer the extensions on both ends. You can try reducing
<ext_thr> if TEtrimmer fails to find full-length TEs. Default: 0.7
--ext_check_win TEXT the check windows size during defining start and end of the consensus
sequence based on the multiple sequence alignment. Used with
<ext_thr>. If <ext_check_win> bp at the end of multiple sequence
alignment has “N” present (ie. positions have similarity proportion
smaller than <ext_thr>), the extension will stop, which defines the
edge of the consensus sequence. Default: 150
--ext_step INTEGER the number of nucleotides to be added to the left and right ends of
the multiple sequence alignment in each extension step. TE_Trimmer
will iteratively add <ext_step> nucleotides until finding the TE
boundary or reaching <max_ext>. Default: 1000
--max_ext INTEGER The maximum extension in nucleotides at both ends of the multiple
sequence alignment. Default: 7000
--gap_thr FLOAT If a single column in the multiple sequence alignment has a gap
proportion larger than <gap_thr> and the proportion of the most
common nucleotide in this column is less than <gap_nul_thr>, this
column will be removed from the consensus. Default: 0.4
--gap_nul_thr FLOAT The nucleotide proportion threshold for keeping the column of the
multiple sequence alignment. Used with the <gap_thr> option. i.e. if
this column has <40% gap and the portion of T (or any other)
nucleotide is >70% in this particular column, this column will be
kept. Default: 0.7
--crop_end_div_thr FLOAT The crop end by divergence function will convert each nucleotide in
the multiple sequence alignment into a proportion value. This
function will iteratively choose a sliding window from each end of
each sequence of the MSA and sum up the proportion numbers in this
window. The cropping will continue until the sum of proportions is
larger than <--crop_end_div_thr>. Cropped nucleotides will be
converted to -. Default: 0.7
--crop_end_div_win INTEGER Window size used for the end-cropping process. Used with the
<--crop_end_div_thr> option. Default: 40
--crop_end_gap_thr FLOAT The crop end by gap function will iteratively choose a sliding window
from each end of each sequence of the MSA and calculate the gap
proportion in this window. The cropping will continue until the sum
of gap proportions is smaller than <--crop_end_gap_thr>. Cropped
nucleotides will be converted to -. Default: 0.1
--crop_end_gap_win INTEGER Define window size used to crop end by gap. Used with the
<--crop_end_gap_thr> option. Default: 250
--start_patterns TEXT LTR elements always start with a conserved sequence pattern.
TEtrimmer searches the beginning of the consensus sequence for these
patterns. If the pattern is not found, TEtrimmer will extend the
search of <--start_patterns> to up to 15 nucleotides from the
beginning of the consensus sequence and redefine the start of the
consensus sequence if the pattern is found. Note: The user can
provide multiple LTR start patterns in a comma-separated list, like:
TG,TA,TC (no spaces; the order of patterns determines the priority
for the search). Default: TG
--end_patterns TEXT LTR elements always end with a conserved sequence pattern. TEtrimmer
searches the end of the consensus sequence for these patterns. If the
pattern is not found, TEtrimmer will extend the search of
<--end_patterns> to up to 15 nucleotides from the end of the
consensus sequence and redefine the end of the consensus sequence if
the pattern is found. Note: The user can provide multiple LTR end
patterns in a comma-separated list, like: CA,TA,GA (no spaces; the
order of patterns determines the priority for the search). Default:
CA
--help Show this message and exit.
The TEtrimmer GUI can also be used to check other TE consensus libraries like the TE library directly from EDTA2, RepeatModeler2, REPET, and other tools.
# Use --help to see all options
python <path_to_GUI_folder>/annoGUI.py --help
# Open your Linux, macOS, or Windows terminal and type
python <path_to_GUI_folder>/annoGUI.py -g <genome.fa> -clib <TE_consensus_library.fa>
# Note: The GUI doesn't perfectly support Windows for "TEAid" plotting function.
# To run the GUI tool, you only need to install BLAST. You do not need to install other packages required by TEtrimmer.

TEtrimmer v1.4.0 Released June.27.2024
TEtrimmer GUI can be used to inspect and improve any TE consensus library. Modified DBSCAN parameter to improve MSA clustering result.
TEtrimmer v1.3.0 Released May.28.2024
Integrated "Extend", "TEAid", and MSA cleaning buttons into TEtrimmer GUI. "TEAid" can generate interactive plots, which can help identifying TE boundaries.
Qian, J., Xue, H., Ou, S., Storer, J., Fürtauer, L., Wildermuth, M. C., Kusch, S., & Panstruga, R. bioRxiv (2024) https://doi.org/10.1101/2024.06.27.600963 TEtrimmer: A novel tool to automate the manual curation of transposable elements.