NLP-based classification of software tools for metagenomics sequencing data analysis into EDAM semantic annotation
by Kaoutar Daoud Hiri, Matjaž Hren and Tomaž Curk
We used machine learning methods to develop a metagenomics tool classification system based on their text description extracted from published papers. The users will quickly and efficiently filter tools of 13 different categories by identifying the tool's specific function.
Motivation: The rapid growth of metagenomics sequencing data makes metagenomics increasingly dependent on computational and statistical methods for fast and efficient analysis. Consequently, novel analysis tools for big-data metagenomics are constantly emerging. One of the biggest challenges for researchers occurs in the analysis planning stage: selecting the most suitable metagenomics software tool to gain valuable insights from sequencing data. The building process of data analysis pipelines is often laborious and time-consuming since it requires a deep and critical understanding of how to apply a particular tool to complete a specified metagenomics task.
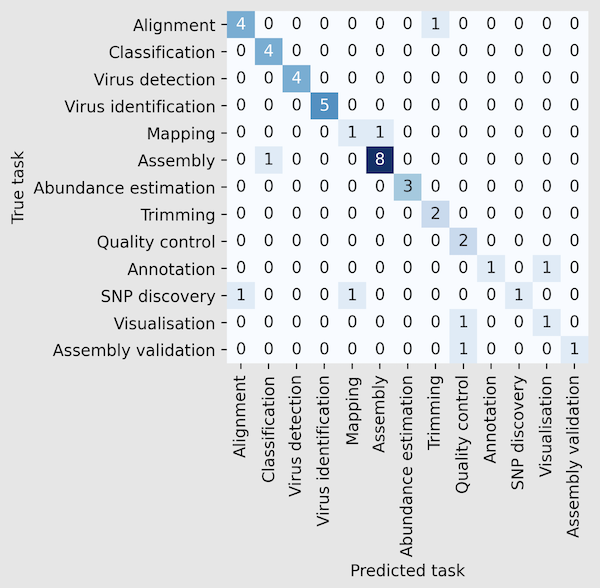
Results: We have addressed this challenge by using machine learning methods to develop a classification system of metagenomics software tools into 13 classes (11 semantic annotations of EDAM and two virus-specific classes) based on the descriptions of the tools. We trained three classifiers (Naive Bayes, Logistic Regression, and Random Forest) using 15 text feature extraction techniques (TF-IDF, GloVe, BERT-based models, and others). The manually curated dataset includes 224 software tools and contains text from the abstract and the methods section of the tools' publications. The best classification performance, with an Area Under the Precision-Recall Curve score of 0.85, is achieved using Logistic regression, BioBERT for text embedding, and text from abstracts only. The proposed system provides accurate and unified identification of metagenomics data analysis tools and tasks, which is a crucial step in the construction of metagenomics data analysis pipelines.
-
-
Create a "ToolName.txt" file containing the publication abstract of the metagenomics tool, and save it in the same folder containing "metagenomics-tool-classifier.py" file.
-
Run the classifier, for example on the file "GAVISUNK.txt":
python metagenomics-tool-classifier.py GAVISUNK.txt
-
Note: In the Jupyter notebooks the names of the EDAM classes are shortened. We keep the part outside the parentheses: (Sequence) alignment, (Taxonomic) classification, (Sequence) assembly, (Sequence) trimming, (Sequencing) quality control, (Sequence) annotation, (Sequence) assembly validation, (RNA-seq quantification for) abundance estimation, SNP-Discovery, Visualization.
-
To explore the code and results, you can execute the Jupyter notebooks individually:
- All source code used to preprocess the text, generate the text embeddings and train the models is in the "Code" folder.
- The data used in this study is in the "Datasets" folder and the results generated by the code are in the "Results" folder.
- The calculations and figure generation are all run inside Jupyter notebooks.
You'll need a working Python environment to run the code. The recommended way to set up your environment is through the Anaconda Python distribution which provides the conda package manager. Anaconda can be installed in your user directory and does not interfere with the system Python installation.
- The anaconda list of packages are in the "NLPclassifier.yaml" file.