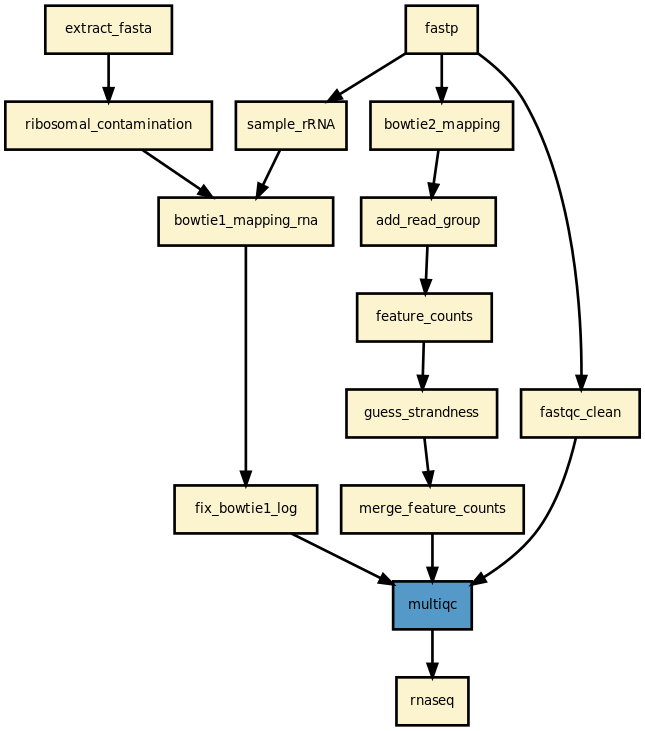
This is is the RNA-seq pipeline from the Sequana project
Overview:
RNASeq analysis from raw data to feature counts
Input:
A set of Fastq Files and genome reference and annotation.
Output:
MultiQC and HTML reports, BAM and bigwig files, feature Counts, script to launch differential analysis
Status:
Production.
Citation(sequana):
Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352
Citation(pipeline):
Installation
sequana_rnaseq is based on Python3, just install the package as follows:
pip install sequana_rnaseq --upgrade
You will need third-party software such as bowtie2/star. However, if you choose to use aptainer/singularity,
then nothing to install except singularity itself ! See below for details.
Usage
sequana_rnaseq --help
sequana_rnaseq --input-directory DATAPATH --genome-directory genome --aligner-choice star
This creates a directory with the pipeline and configuration file. You will then need
to execute the pipeline:
cd rnaseq
sh rnaseq.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can
retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
This pipelines requires lots of third-party executable(s). Here is a list that
may change. A Message will inform you would you be missing an executable:
bowtie
bowtie2>=2.4.2
STAR
featureCounts (subread package)
picard
multiqc
samtools
Note that bowtie>=2.4.2 is set to ensure the pipeline can be used with python 3.7-3.8-3.9 and the sequana-wrappers that supports bowtie2 with option --threads only (not previous versions). See environment.yaml or conda.yaml for latest list of required third-party tools.
Or use the conda.yaml file available in this repository. If you start a new
environment from scratch, those commands will create the environment and install
all dependencies for you:
For Linux users, we provide singularity images available through within the damona project (https://damona.readthedocs.io).
Details
This pipeline runs a RNA-seq analysis of sequencing data. It runs in
parallel on a set of input FastQ files (paired or not).
A brief HTML report is produced together with a MultiQC report.
This pipeline is complex and requires some expertise for the interpretation.
Many online-resources are available and should help you deciphering the output.
Yet, it should be quite straigtforward to execute it as shown above. The
pipeline uses bowtie1 to look for ribosomal contamination (rRNA). Then,
it cleans the data with cutapdat if you say so (your data may already be
pre-processed). If no adapters are provided (default), reads are
trimmed for low quality bases only. Then, mapping is performed with standard mappers such as
star or bowtie2 (--aligner option). Finally,
feature counts are extracted from the previously generated BAM files. We guess
the strand and save the feature counts into the directoy
./rnadiff/feature_counts.
The pipelines stops there. However, RNA-seq analysis are followed by a different
analysis (DGE hereafter). Although the DGE is not part of the pipeline, you can
performed it with standard tools using the data in ./rnadiff directory. One such
tool is provided within our framework (based on the well known DEseq2 software).
where ANNOT is the annotation file of your analysis, FEAT and ATTR the attribute
and feature used in your analysis (coming from the annotation file).
This produces a HTML repot summarizing you differential analysis.
Rules and configuration details
Here is the latest documented configuration file
to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
Warning
the RNAseQC rule is switch off and is not currently functional in
version 0.9.X
Issues
In the context of eukaryotes, you will need 32G of memory most probably. If this is too much,
you can try to restrict the memory. Check out the config.yaml file in the star section.
Changelog
Version
Description
0.20.0
Fix regression due to new sequana version
Update summary html to use new sequana plots
0.19.3
fix regression with click to set the default rRNA to 'rRNA' again.
0.19.2
fix bowtie1 regression in the log file, paired end case in
multiqc and rnadiff script (regression)
set genome directory default to None to enforce its usage
0.19.1
add rnaseqc container.
Update rseqc rules (redirection)
cleanup onsuccess rule
0.19.0
Refactorisation to use click
0.18.1
fastp multiqc regression. Fixed missing sample names by updating
multiqc_config and adding sample names in the output filename
0.18.0
New plots in the HTML reports. Includes version of executables.
0.17.2
CHANGES: in star section, added --limitBAMsortRAM and set to 30G
BUG: Fix missing params (options) in star_mapping rule not taken
into account
0.17.1
use new rulegraph / graphviz apptainer
0.17.0
fastp step changed to use sequana-wrappers. Slight change in
config file. The reverse and forward adapter options called
rev and fwd have been dropped in favor of a single adapters option.
v0.17.0 config and schema are not compatible with previous
versions.
Update singularity containers and add new one for fastp
0.16.1
fix bug in feature counts automatic strand balance detection. Was
always using the stranded case (2).
add singularity workflow for testing
fix documentation in config.yaml
0.16.0
star, salmon, bam_coverage are now in sequana wrappers, updated
the pipeline accordingly
updated config file and schema to include resources inside the
config file (so as to use new --profile option)
set singularity images in all rules
star wrappers has changed significantly to use star
recommandation. To keep using previous way, a legacy option
is available and set to True in this version.
bamCoverage renamed in bam_coverage in the config file
multiqc_config removed redundant information and ordered
the output in a coherent way (QC and then analysis)
0.15.2
Fix bowtie2 rule to use new wrappers. Use wrappers in
add_read_group and mark_duplicates
When initiating the pipeline, provide information about the GFF
mark duplicates off by default
feature_counts has more options in the help. split options into
feature/attribute/extra_attributes.
HTML reports better strand picture and information about rRNA
refactorising the main standalone and config file to split feature
counts optiions into feature and attribute. Sanoty checks are ow
provided (--feature-counts-attribute, --feature-counts-feature-type)
can provide a custom GFF not in the genome directory
can provide several feature from the GFF. Then, a custom GFF is
created and used
fix the --do-igvtools and --do-bam-coverage with better doc
0.10.0
9/12/2020
Fixed bug in sequana/star_indexing for small genomes (v0.9.7).
Changed the rnaseq requirements to benefit from this bug-fix that
could lead to seg fault with star aligner for small genomes.
Report improved with strand guess and plot
0.9.20
7/12/2020
BUG in sequana/star rules v0.9.6. Fixed in this release.
In config file, bowtie section 'do' option is removed. This is now
set automatically if rRNA_feature or rRNA_file is provided. This
allows us to skip the rRNA mapping entirely if needed.
fastq_screen should be functional. Default behaviour is off. If
set only phiX174 will be search for. Users should build their own
configuration file.
star/bowtie1/bowtie2 have now their own sub-directories in the
genome directory.
added --run option to start pipeline automatically (if you know
what you are doing)
rnadiff option has now a default value (one_factor)
add strandness plot in the HTML summary page
0.9.19
Remove the try/except around tolerance (guess of strandness) to
make sure this is provided by the user. Final onsuccess benefits
from faster GFF function (sequana 0.9.4)
0.9.18
Fix typo (regression bug) + add tolerance in schema + generic
title in multiqc_config. (oct 2020)
0.9.17
add the tolerance parameter in the feature_counts rule as a user
parameter (config and pipeline).
0.9.16
Best feature_counts is now saved into rnadiff/feature_counts
directory and rnadiff scripts have been updated accordingly
the most probable feature count option is now computed more
effectivily and incorporated inside the Snakemake pipeline (not in
the onsuccess) so that multiqc picks the best one (not the 3
results)
the target.txt file can be generated inside the pipeline if user
fill the rnadiff/conditions section in the config file
indexing options are filled automatically when calling
sequana_rnaseq based on the presence/absence of the index
of the aligner being used.
salmon now integrated and feature counts created (still WIP in
sequana)
0.9.15
FastQC on raw data skipped by default (FastQC
for processed data is still available)
Added paired options (-p) for featureCounts
Switch back markduplicates to False for now.
0.9.14
Use only R1 with bowtie1
set the memory requirements for mark_duplicates in cluster_config
file
Set temporary directory for mark_duplicates to be local ./tmp
0.9.13
set mark_duplicate to true by default
use new sequana pipeline manager
export all features counts in a single file
custom HTML report
faster --help calls
--from-project option added
0.9.12
include salmon tool as an alternative to star/bowtie2
include rnadiff directory with required input for Differential
analysis
0.9.11
Automatic guessing of the strandness of the experiment
0.9.10
Fix multiqc for RNAseQC rule
0.9.9
Fix RNAseQC rule, which is now available.
Fix ability to use existing rRNA file as input
0.9.8
Fix indexing for bowtie1 to not be done if aligner is different
add new options: --feature-counts-options and --do-rnaseq-qc,
--rRNA-feature
Based on the input GFF, we now check the validity of the rRNA
feature and feature counts options to check whether the feature
exists in the GFF
schema is now used to check the config file values
add a data test for testing and documentation
0.9.7
fix typo found in version 0.9.6
0.9.6
Fixed empty read tag in the configuration file
Possiblity to switch off cutadapt section
Fixing bowtie2 rule in sequana and update the pipeline accordingly
Include a schema file
output-directory parameter renamed into output_directory (multiqc
section)
handle stdout correctly in fastqc, bowtie1, bowtie2 rules
if a fastq_screen.conf is provided, we switch the fastqc_screen
section ON automatically
0.9.0
Major refactorisation.
remove sartools, kraken rules.
Indexing is now optional and can be set in the configuration.
Configuration file is simplified with a general section to enter
the genome location and aligner.
Fixed rules in sequana (0.8.0) that were not up-to-date with
several executables used in the pipeline including picard,
fastq_screen, etc. See Sequana Changelog for details with respect
to rules changes.
Copying the feature counts in main directory ready to use for
a differential analysis.