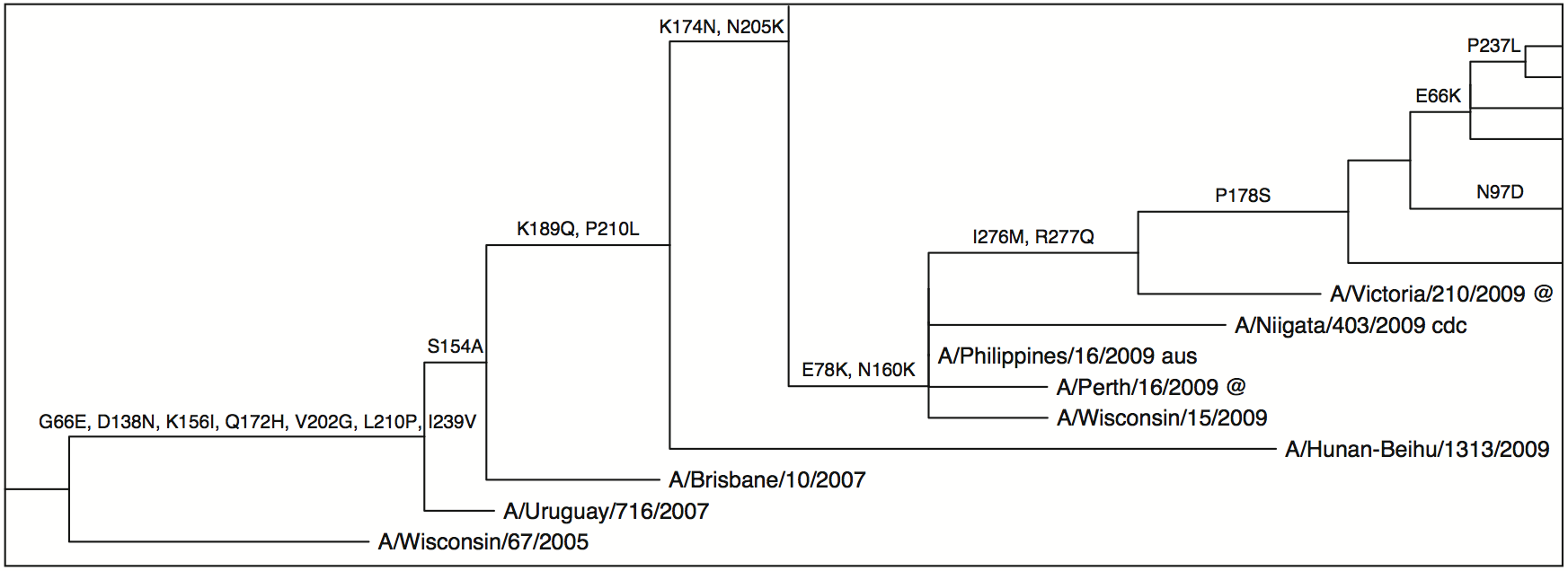
This little tool glues together several other tools to allow a user to input a codon alignment in FASTA format and produce an annotated phylogenetic tree showing which substitutions occurred on a given branch. It uses the following tools and libraries:
- RAxML is a program for sequential and parallel Maximum Likelihood based inference of large phylogenetic trees [1].
- PAML is a suite of software tools for phylogenetic analysis [2]. We use it to estimate the branch lengths and infer the ancestral sequences.
- FigTree is a graphical viewer of phylogenetic trees. The annotated tree can be opened in Figtree and the tree can be prepared (as close as is possible) as you would like for printing or further editing in a program like Adobe Illustrator.
- Java libraries PAL [3] and BioJava [4], among others, are used to read, manipulate and write alignment files and phylogenetic trees.

This application have been confirmed to work on Mac OS 10.6+ and Linux. You need to download and install:
- RAxML, which can be downloaded from Alexis Stamatakis' lab homepage.
- PAML, which can be downloaded from Ziheng Yang's homepage.
- A recent Java runtime (1.6+).
- Figtree, which can be downloaded from Andrew Rambaut's homepage. The output of the treesub program is designed to work with Figtree but other tools might work as well.
The process has been designed to be as straightfoward as possible. However, we describe all the steps here to clarify the workflow. In theory, you only need to provide a sequence alignment (step 1 and 2) and hit 'RUN'.
-
Prepare the sequences and alignment. The alignment must be a coding sequence alignment without any STOP codons. It is recommended that you remove any signal or trailing peptides, otherwise these will throw off the numbering of sites. The alignment must be in FASTA format. The first sequence in the alignment (i.e. the one at the top of the FASTA file) is used as your outgroup sequence. There is no requirement for any specific labeling. However, it is recommended that you avoid commas or parenthesis in your FASTA labels as these can cause issues when reading and writing tree files.
-
Create a new directory for each analysis. As we run several different programs that produce many different files, some with the same names, it is recommended that you create a new (i.e. empty) directory for each run. Put your FASTA alignment into this new directory.
-
Building the tree and ancestral reconstruction (RAxML & PAML). You don't necessarily need to know all these details; they are provided here for your information and to assist with any troubleshooting. Once you have saved your alignment in a new directory, start the treesub program by double-clicking on treesub.jar. The first time you run the program, you will need to supply the path to the RAxML and PAML programs. Use the 'Browse...' buttons to select the raxmlHPC and baseml executables. These are saved and you won't need to enter them for subsequent runs. Finally, select your FASTA alignment using the 'Browse...' button and click 'Run'.
- You can name your FASTA file as you like. However, before we start any processing, we rename the FASTA alignment to a file named
alignment. We do this because all the subsequent programs will expect a file with this name. - We run a small utility program that reads the FASTA alignment and produces (a) a file called
alignment.names, which is a list of all the labels for your sequences and (b) a PHYLIP formatted alignment calledalignment.raxml.phylip. PHYLIP formatted alignments are required by RAxML and PAML. Additionally, the sequences are renamed to 'seq_1', 'seq_2', 'seq_3' and so on. This is to ensure that there are no name conflicts or errors in accepted name format in the subsequent programs. - RAxML is run with
alignment.raxml.phylipto estimate the tree topology by maximum likelihood. We use the GTRGAMMA model. - A small utility then roots the resulting RAxML tree by your outgroup sequence, which should have been the first sequence in your original FASTA alignment file (hence, we know this as 'seq_1'). This is required to make sure that the ancestral reconstruction goes in the right direction.
- We run the PAML program
basemlto estimate the branch lengths and perform the ancestral reconstruction.
- You can name your FASTA file as you like. However, before we start any processing, we rename the FASTA alignment to a file named
-
Producing the annotated tree. Once we have run RAxML and PAML, the program is ready to produce the annotated tree file. It will read the optimised trees and ancestral substitutions from the PAML output and the sequence names from the
alignment.namesfile and write a new tree calledsubstitutions.tree. It will also create a table of all synonymous and non-synonymous substitutions in a tab-separated values files calledsubstitutions.tsv. It specifies the node/branch number, codon change and amino acid change for the substitutions. This can be opened in a text editor or a program like Excel. You may find it useful to take a look at the list of your sequences inalignment.names. Each line has the name of one sequence. You can edit the names of your sequences in this file. The only hard rules are to keep the same order of sequences and to avoid using commas and parentheses. For example, you could rename "A-URUGUAY-716-2007" to "A/Uruguay/716/2007 cdc @". You may find it easier to do this kind of renaming in this file rather than in a tree viewing program or graphics editing program. If you make changes to the alignment.names and save the file (it must keep the same name), you can generate a new tree file by clicking the 'Re-annotate' button. Note that you do not have to do this re-annotating at the time of your initial analysis. Simply open the Annotator program (by double-clicking on treesub.jar), select the same alignment file (in the same directory, with all the miscellaneous results files) and click 'Re-annotate'. -
Viewing the annotated tree. Start the Figtree viewing program and open the
substitutions.treefile. There are many options in Figtree to manipulate the rendering of the tree. The following are an example of the types of things you can do using the panels in the sidebar:- In the 'Layout' panel, move the 'Expansion' slider to spread out the taxa so the labels do not overlap.
- In the 'Trees' panel, tick the 'Order nodes' checkbox and set ordering to 'decreasing'.
- The 'Tip Labels' panel allows you to choose what to display as the taxa name: the names, all substitutions, non-synonymous substitutions, node number, or 'FULL', which displays the node number, taxon name and non-synonymous substitutions together.
- Tick the 'Node Labels' panel and in the panel select 'NUMBER' for display. This will show the internal node numbers which are needed to decipher the list of substitutions in the 'substitutions.tsv' file.
- Tick the 'Branch Labels' panel and in the panel selection 'NONSYNSUBS' for display. This shows any non-synonymous substitutions that occurred along the branch.
- Finally, you can use the 'Layout' panel again to spread out the tree to avoid overlapping labels by using the 'Zoom' and 'Expansion' sliders.
Each time you have the tree as you like, you can export to PDF or a graphics image. If you export to PDF (or EPS) you can edit the result in Adobe Illustrator (or Inkscape).

An example of entries in the 'substitutions.tsv' file:
| branch | site | codon_from | codon_to | aa_from | aa_to | string | non-syn? |
|---|---|---|---|---|---|---|---|
| 3 | 108 | AAA | CAA | K | Q | K108Q | Y |
| 3 | 237 | CCC | CTC | P | L | P237L | Y |
| 207 | 16 | GCT | Gcc | A | A | A16A | N |
| 30 | 345 | AGA | AAA | R | K | R345K | Y |
etc.
If you already have run PAML analysis, you can run the following to create substitutions.tree and substitutions.tsv files. You need to have run PAML with the appropriate options. For example, if the rst file is in /path/to/paml/results run:
java -cp dist/treesub.jar treesub.ancestral.ParseRST /path/to/paml/results
Thanks to Mario dos Reis, John McCauley and Vicki Gregory for feedback and ideas for improvement.
- Stamatakis, A. 2006. RAxML-VI-HPC: Maximum Likelihood-based Phylogenetic Analyses with Thousands of Taxa and Mixed Models. Bioinformatics 22(21):2688–2690.
- Yang, Z. 2007. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Molecular Biology and Evolution 24: 1586-1591.
- Drummond, A., and K. Strimmer. 2001. PAL: An object-oriented programming library for molecular evolution and phylogenetics. Bioinformatics 17: 662-663.
- R.C.G. Holland; T. Down; M. Pocock; A. Prlić; D. Huen; K. James; S. Foisy; A. Dräger; A. Yates; M. Heuer; M.J. Schreiber. 2008. BioJava: an Open-Source Framework for Bioinformatics. Bioinformatics 24 (18): 2096-2097.