
We herewith introduce EMCIP, an Ensemble Model for Cdr1 Inhibitor Prediction, featured in our paper:
Trinh, T.-C., Falson, P., Tran-Nguyen, V.-K.* & Boumendjel, A.* Ligand-Based Drug Discovery Leveraging Traditional Machine Learning and Deep Learning Methodologies Exemplified by Prediction of Cdr1 Inhibitors. (2024)
To set up the environment for EMCIP, you will need Conda (v.24.1.2). For Conda installation instructions, refer to this link.
Run the following commands in your terminal to install the GUI for EMCIP:
git clone https://github.com/trinhthechuong/Cdr1_inhibitors.git
cd Cdr1_inhibitors
conda env create --file environment.yml
conda activate EMCIP_env
pip install -r requirements.txt
streamlit run EMCIP.pyEMCIP provides two functionalities: Batch Prediction and Molecule Prediction.
In the main menu, select the Predict a batch option and follow these steps:
- Upload a *.csv file. The file must contain two columns: the first for molecule names or IDs, and the second for the SMILES of these molecules. Then, provide names for your columns and choose the number of processors for the calculation.
- Click the
Featurizebutton and wait for the process to complete. During this step, your molecules will be standardized and converted into various molecular representations (RDK5, RDK6, RDK7, Avalon, Mordred, Gobbi Pharmacophore) and 3D molecular graphs. - Once featurization is complete, click the
Predictionbutton to predict your data.
All featurized datasets and prediction results are saved in the Cdr1_classification folder. Click the Restart button to start predicting another file.

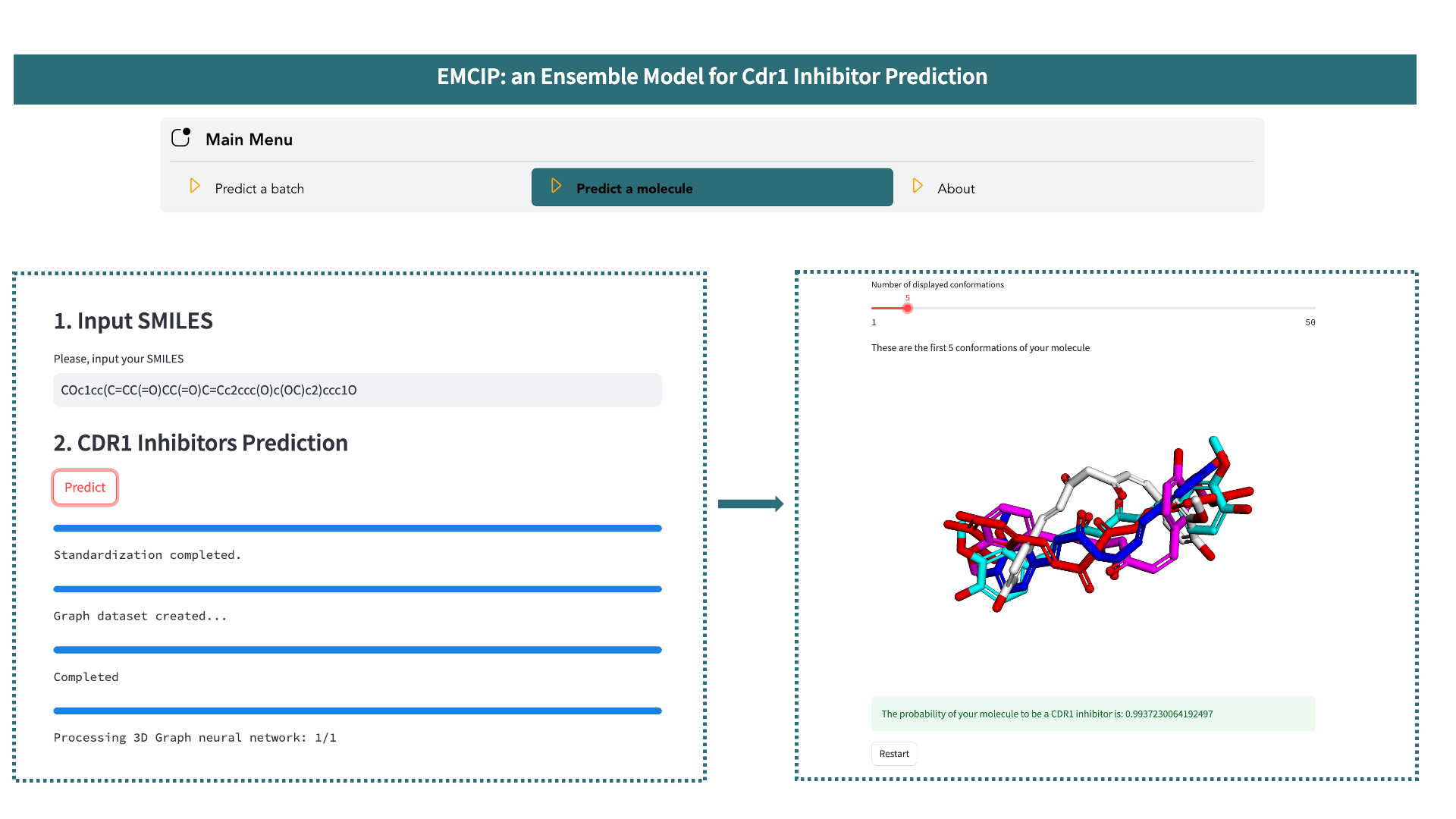
In the main menu, select the Predict a molecule option and follow these steps:
- Enter the SMILES string of your molecule.
- Click the
Predictbutton and wait for the completion of the process. During this step, your molecule will be standardized, converted into various molecular representations (RDK5, RDK6, RDK7, Avalon, Mordred, Gobbi Pharmacophore) and 3D molecular graphs, before being evaluated by the EMCIP model for prediction. - The output is the predicted probability of your molecule being a Cdr1 inhibitor.
Additionally, you can interact with the generated conformations used as input for MIL-3D-GNN.

The EMCIP model is also available for direct prediction on the Hugging Face platform EMCIP-Hugging Face. However, for optimal performance, we recommend installing EMCIP locally to leverage the power of your local processors.
You can find the instructional video on how to use our model here.
The dataset folder stores all training data and corresponding results.
- original_dataset.csv: This file contains all assembled molecules for EMCIP along with their references.
Featurized_datafolder:BM_stratified_sampling: This sub-folder stores all datasets used for training (training set), validation (external test set, and hard test set).MIL_3D_GNN: This sub-folder stores graph datasets specifically used for the MIL-3D-GNN model.
molecular_representation_analysissub-folder contains all 16 ligand-based structural representation datasets and the results of the associated meta-analysis, including Wilcoxon signed-rank test.
ml_model_selectionsub-folder stores all validation results, including Bemis-Murcko Scaffold 5-fold cross-validation and external test set validation of traditional machine learning models.bayesian_estimationsub-folder houses the results comparing machine learning model performance through Bayesian estimation
- Validation results for MIL-3D-GNN on validation, external, and hard test sets are stored in the
validation_mil_3d_gnnsubfolder. - To view the hyperparameter tuning process for MIL-3D-GNN on mlflow, run the following commands in your terminal:
cd MIL_3D_GNN
mlflow server --host 127.0.0.1 --port 8080- graph_featurization.ipynb: This Jupyter Notebook details the process of converting molecules into graph representations for use with the MIL-3D-GNN model.
For further queries, please contact:
- The-Chuong Trinh: the-chuong.trinh@etu.univ-grenoble-alpes.fr, thechuong123@gmail.com
- Dr. Viet-Khoa Tran-Nguyen: viet-khoa.tran-nguyen@u-paris.fr, khoatnv1993@gmail.com
- Pr. Achène Boumendjel: ahcene.boumendjel@univ-grenoble-alpes.fr