Cloud-optimized primary analysis pipelines for RNA-seq, miRNA-seq, ChIP-seq, RNAEditing and variant calling in whole-genome/whole-exome DNA-Seq.
Bionformatic analysis of large-scale next-generation sequencing (NGS) data requires significant compute resources. While cloud computing makes such processing power available on-demand, the administration of dynamic compute clusters is a daunting task for most working biologists. To address this pain point, the Center for Computational Biology and Bioinformatics at the University of California, San Diego, has developed Cirrus-NGS, a turn-key solution for common NGS analyses using Amazon Web Services (AWS).
Cirrus-NGS provides primary analysis pipelines for RNA-Seq, miRNA-Seq, ChIP-Seq, RNAEditing and whole-genome/whole-exome sequencing data. Cirrus users need not have any bioinformatics tools for these pipelines installed on their local machine, as all computation is performed using AWS clusters and all results are uploaded to AWS's S3 remote storage. The clusters are created dynamically through Cirrus-NGS, based on a custom machine image with all the necessary software pre-installed. Users manage clusters and run pipelines from within a web browser, using flexible but light-weight Jupyter notebooks.
Because Cirrus-NGS employs AWS for computation and storage, users must have an active AWS account. If you do not have such an account, visit https://docs.aws.amazon.com/AWSEC2/latest/UserGuide/get-set-up-for-amazon-ec2.html for guidance on how to create one, and execute the steps it describes.
Since release v1.2, Cirrus-NGS is provided docker installation, users just run the below commands to pull the latest image from docker and then launch the jupiter notebook to run Cirrus-NGS.
docker pull g1xu/cirrusngs:latest
Then run the below command to create a container from the image, and start the new container or you can stop it after you finish analysis.
docker run -d -p 8888:8888 -it g1xu/cirrusngs bash
docker start <container_id>
docker stop <container_id>
The below command allows to login the container.
docker exec -it <container_id> bash
After users login to the container, you need to go to the directory /mnt/workspace to run the below command to launch a Jupiter notebook server.
/mnt/workspace/software/anaconda3/bin/jupyter notebook --allow-root --ip 0.0.0.0 --no-browser --port=8888
Finally, the Jupiter notebook server is running and you can use Cirrus-NGS.
While Cirrus-NGS itself is provied through github, its supporting libraries are best installed through conda, a cross-platform package manager. If you do not have conda installed already, download the appropriate Python-3.6-based installer from the links below:
- linux: https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh
- OSX: https://repo.continuum.io/miniconda/Miniconda3-latest-MacOSX-x86_64.sh
Then run the installer with the following script, replacing <miniconda_py3.sh> with the name of the install script downloaded above:
bash <miniconda_py3.sh> -b -p $HOME/miniconda3
echo "export PATH=\"$HOME/miniconda3/bin:\$PATH\"" >>$HOME/.bashrc
source $HOME/.bashrc
Cirrus-NGS is available for linux-64 or osx-64 platforms, and requires python 3.5 or higher.
In the directory in which you would like to install Cirrus-NGS, run the following commands:
conda install paramiko pyyaml git jupyter notebook
pip install aws-parallelcluster
git clone https://github.com/ucsd-ccbb/Cirrus-NGS.git
These commands install the necessary supporting libraries and create a new directory called Cirrus-NGS that holds the Cirrus software.
Since the Cirrus-NGS interface is provided through Jupyter notebooks, the first step of running Cirrus is to start a local Jupyter notebook server. From the Cirrus-NGS directory, run:
cd notebooks/cirrus-ngs
jupyter notebook
You will receive a message like
[I 10:48:39.779 NotebookApp] Serving notebooks from local directory: /Users/<YourName>/Cirrus-NGS/notebooks
[I 10:48:39.779 NotebookApp] 0 active kernels
[I 10:48:39.779 NotebookApp] The Jupyter Notebook is running at: http://localhost:8889/?token=8dcb989e6852e3dfd679307470e17696c32771432c881573
[I 10:48:39.779 NotebookApp] Use Control-C to stop this server and shut down all kernels (twice to skip confirmation).
Note the URL at which the server is running. It is usually http://localhost:8888, but if your system already has something running on port 8888, it may default so something else (e.g., in the example above, it is http://localhost:8889).
Do not shut down the Jupyter notebook server or close the terminal window in which it is running until you are done using Cirrus-NGS, as it must be running in order for the Cirrus-NGS notebooks to function. When you are done using Cirrus and ready to shut down the notebook server, simply type Control-C in the terminal window where it is running and follow the prompts.
A user begins by defining a compute cluster. After this, sh/e decides upon the pipeline to run, which determines the exact inputs required, and then creates (in a spreadsheet program or text editor) a design file that specifies the details of the input sequencing data. S/he starts the Jupyter notebook for the chosen pipeline and inputs the path to the design file and other required inputs (such as AWS credentials, etc), then executes the notebook code to start the pipeline processing.
The notebook creates a yaml file summarizing all of the user input and transfers that file to the cluster, where it directs cluster-native code to sequentially execute the analysis steps specified by the user in a distributed fashion. Upon completion of each step, the output is uploaded to the user's specified S3 output bucket, from which it can be accessed at any point. During processing, the user can check the status of the pipeline run from within the notebook.
A Note About Terminology: Throughout, the term "pipeline" is used to describe functionality for analyzing a general type of sequencing (such as RNA-Seq or whole-genome sequencing). Each pipeline can have multiple, more granular "workflows" that support different approaches to processing the same type of sequencing data; for instance, the RNA-Seq pipeline contains four different workflows (star_gatk, star_htseq, star_rsem, and kallisto).
The following pieces of information are required by all functionality in Cirrus-NGS:
- The AWS access key ID for your AWS account
- See https://docs.aws.amazon.com/general/latest/gr/managing-aws-access-keys.html to find your existing access key or create a new one)
- The AWS secret access key for your AWS account (see above link for more information)
- A valid key pair (.pem) file for your AWS account
- If you encounter a
Permission denied (publickey) error, note that the permissions on your key (.pem) file must be set so that it is not public, e.g. by runningchmod 0400 <mypemfilename>.pemin the directory where the .pem file is located. - See https://docs.aws.amazon.com/AWSEC2/latest/UserGuide/ec2-key-pairs.html#having-ec2-create-your-key-pair for further details.
- If you encounter a
Additional inputs are required for specific actions, and are discussed in the relevant sections below.
Run the following steps to define a compute cluster. You may then use that cluster for all Cirrus-NGS work going forward without having to rerun these steps (although you may also rerun them to create additional clusters, if desired).
-
Gather the required settings information; in addition to the valued described above in the Gathering Required Inputs section, you will need:
-
The AWS region in which you want to run your cluster
- A table of available region values is shown at https://docs.aws.amazon.com/AWSEC2/latest/UserGuide/using-regions-availability-zones.html#concepts-available-regions. Note that different regions charge different prices for computing, so do not choose one blindly. In general, you will want to select the closest region to you geographically that has an acceptable price for the virtual computing nodes (called "instances") that you wish to run (see below for more details).
-
The instance type to use for the master node of your cluster
- This node runs as long as the cluster is active, and directs the other nodes; because of its central role, it will always be run with AWS's standard, "on-demand" pricing.
- Visit https://aws.amazon.com/ec2/pricing/on-demand/ and select a region relevant to you in the "Region" dropdown box in order to to view the per-hour prices for available instance types in that region.
- Generally speaking, the master node will need to run on an instance with at least 2 vCPUs and at least 8 GiB of memory (such as an
m4.large).
-
The instance type to use for the compute nodes of your cluster
- These nodes are dynamically added to and removed from the cluster to accommodate the amount of calculation the cluster is asked to do.
- Generally speaking, the compute nodes will need to have CPU and memory capacity greater than or equal to that of the master node.
- These nodes are run with AWS's discounted "spot" pricing (see below for details).
-
The spot price for the compute nodes of your cluster
- If AWS has left-over capacity of a particular instance type after filling all on-demand requests for that type, it sells the remaining capacity at a dynamically-set lower price; the "spot price" you specify is the maximum hourly cost, in US dollars, that you are willing to pay for each such instance (see https://aws.amazon.com/ec2/faqs/ for details).
- The going spot rate for a particular instance type in a particular region is constantly changing bsaed on demand. Visit https://aws.amazon.com/ec2/spot/pricing/ (remember to select the correct region for you, as described above!) to view the current spot prices for instances available in your region.
- If the going spot rate for your chosen compute instance type goes above your specified spot price, you will not be charged more, but your compute nodes will shut down without warning as they are re-allocated to another user. Therefore, it is wise to choose a spot price that represents the maximum you are willing to pay, rather than under-bidding.
-
The number of compute nodes in your cluster when it is first started
- If you do not have a good idea of how many you will need, it is fine to use zero by default.
-
The size in GiB of the virtual storage disk for the cluster
-
The VPC (Virtual Private Cloud) id for your AWS account
- If you do not know the VPC id, you can find it by going part-way through the process of launching an AWS compute instance. Follow the instructions at https://docs.aws.amazon.com/AWSEC2/latest/UserGuide/launching-instance.html up until step 6 (it does not matter what values you select during these steps, as you won't need to actually launch the instance). Step 6 brings you to the "Configure Instance Details" screen, on which you can find your VPC id, as well as your subnet ids (see next item):
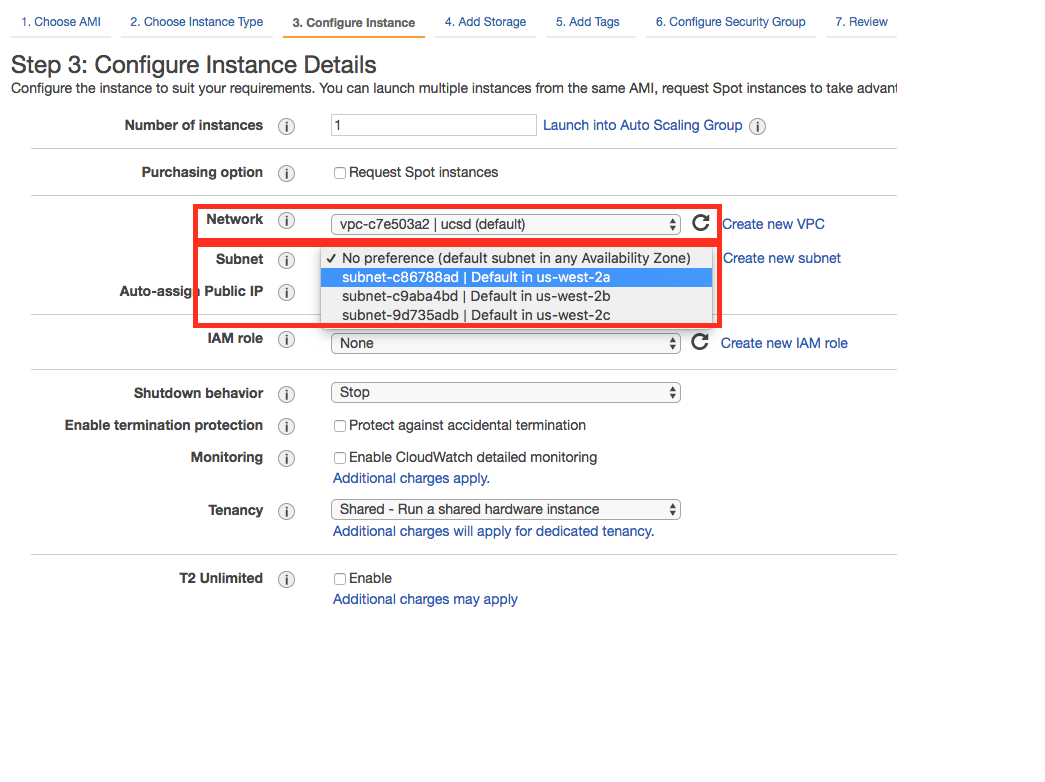
-
The subnet id for your AWS account for the region in which you wish to create the cluster (see above for how to find this value)
-
-
In your browser, visit the address at which the Jupyter notebook server is running
- This is usually http://localhost:8888, but see the Starting the Notebook Server section for additional details.
- You will see a list of all the Cirrus-NGS notebooks:
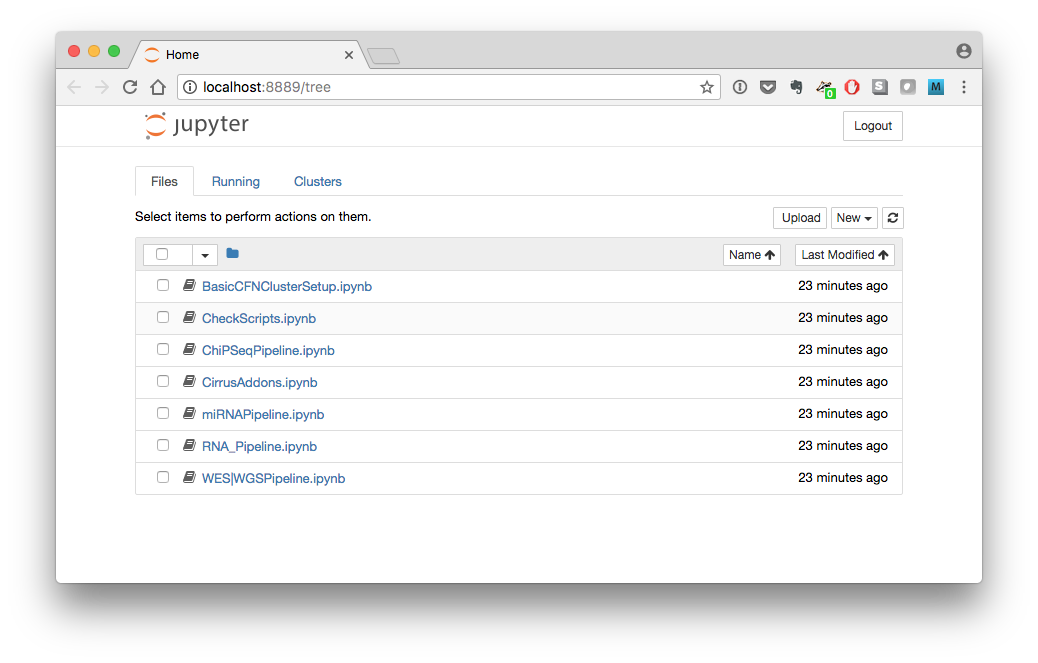
-
Click on the
BasicAWSClusterSetup.ipynbnotebook to start it. -
Fill in the parameters in the first two cells in the notebook, using the values identified above in the Gathering Required Inputs section where relevant.
-
Run the notebook.


-
Choose the pipeline you wish to run.
- Cirrus-NGS currently offers pipelines for RNA-Seq, miRNA-seq, ChIP-seq, RNAEditing and variant calling in whole-genome/whole-exome DNA-Seq.
-
Create a design file for your data.
- The design file is a tab-separated text file with either two or three columns (depending on the workflow chosen) that specifies the names of the sequence files to process and the necessary metadata describing them.
- See the Building a Design File section below for full specifications of the design file format.
-
Gather the required settings information; in addition to the valued described above in the Gathering Required Inputs section, you will need:
- The URL of an existing AWS S3 remote storage bucket where you have placed your sequencing data.
- Visit https://docs.aws.amazon.com/AmazonS3/latest/gsg/GetStartedWithS3.html and execute the "Sign Up for Amazon S3" and "Create a Bucket" steps (click on the boxes in the workflow diagram for detailed instructions) if you do not already have a bucket set up.
- To place your sequencing files into the bucket, follow the directions for the "Add an Object to a Bucket" step, or use an S3-enabled transfer client like Cyberduck.
- (Optional) The URL of a second existing AWS S3 remote storage bucket for Cirrus-NGS outputs.
- The URL of an existing AWS S3 remote storage bucket where you have placed your sequencing data.
-
In your browser, visit the address at which the Jupyter notebook server is running
- This is usually http://localhost:8888, but see the Starting the Notebook Server section for additional details.
- You will see a list of all the Cirrus-NGS notebooks:
-
Click on the appropriate notebook for your chosen pipeline to start it.
-
Fill in the parameters in the first two cells in the notebook, using the values identified above in the Gathering Required Inputs section where relevant.
-
Run the notebook.
-
After the pipeline processing is complete, download the output files from your S3 bucket for further analysis locally.
- To download the outputs from your bucket, follow the directions for the "View an Object" step at https://docs.aws.amazon.com/AmazonS3/latest/gsg/OpeningAnObject.html or use an S3-enabled transfer client like Cyberduck.
The design file is the primary user input to Cirrus-NGS. It is a tab-separated text file that specifies the names of the sequence files to process and the necessary metadata describing them. For the RNA-Seq and miRNA-Seq pipelines, it contains two columns, while for the ChIP-Seq and whole-exome/whole-genome sequencing pipelines, it contains three columns (of which the first two are the same as in the two-column format). The design file has no header line.
In the two-column format, the first column is the filename of the sample (with extensions: e.g. fastq.gz), while the second column is the name of the group associated with that sample. (Group names are up to the user, but they are generally set to experimental conditions. For example, all control samples can be given a group named "control".)
For example, a two-column design file for two single-end-sequenced samples named mysample1 and mysample2 might look like:
mysample1.fastq.gz groupA
mysample2.fastq.gz groupB
If the sequencing data is paired-end, the first column includes the name of the forward sequencing file, followed by a comma, followed by the name of the reverse sequencing file (note that there must not be any spaces between these two file names--only a comma!) An example two-column design file for two paired-end-sequenced samples named mysample1 and mysample2 might look like:
mysample1_fwd.fastq.gz,mysample1_rev.fastq.gz groupA
mysample2_fwd.fastq.gz,mysample2_rev.fastq.gz groupB
The three-column format has the same first two columns as the two-column format, but adds a third column containing a pipeline-specific identifier that differentiates sample types for the ChIP-Seq and whole-genome/whole-exome sequencing pipelines.
- For the ChIP-Seq pipeline, each sample is identified as either Chip or Input.
- For the whole-genome/whole-exome sequencing pipeline, each sample is identified as either Tumor or Normal.
The following constraints apply to three-column design files:
-
The sample type identifiers are case-sensitive.
- For example, a three-column design file for two paired-end-sequenced ChIP-Seq samples named
mysample1andmysample2might look like
mysample1_fwd.fastq.gz,mysample1_rev.fastq.gz groupA Chip mysample2_fwd.fastq.gz,mysample2_rev.fastq.gz groupB Input - For example, a three-column design file for two paired-end-sequenced ChIP-Seq samples named
-
Each three-column design file must use either Chip and Input sample type identifiers or Normal and Tumor sample type identifiers, but not both.
- A design file like the example below would thus be invalid:
mysample1_fwd.fastq.gz,mysample1_rev.fastq.gz badExample1 Chip mysample2_fwd.fastq.gz,mysample2_rev.fastq.gz badExample1 Tumor -
Each group must have exactly one sample designed with each of the two relevant sample type identifiers. * A design file like the example below would thus be invalid:
mysample1_fwd.fastq.gz,mysample1_rev.fastq.gz badExample2 Normal mysample2_fwd.fastq.gz,mysample2_rev.fastq.gz badExample2 Normal -
If two samples are designated as forming a Chip/Input or Normal/Tumor pair, they must both have the same group.
- A design file like the example below would thus be invalid:
mysample1_fwd.fastq.gz,mysample1_rev.fastq.gz badExample3 Normal mysample2_fwd.fastq.gz,mysample2_rev.fastq.gz badExample4 Tumor