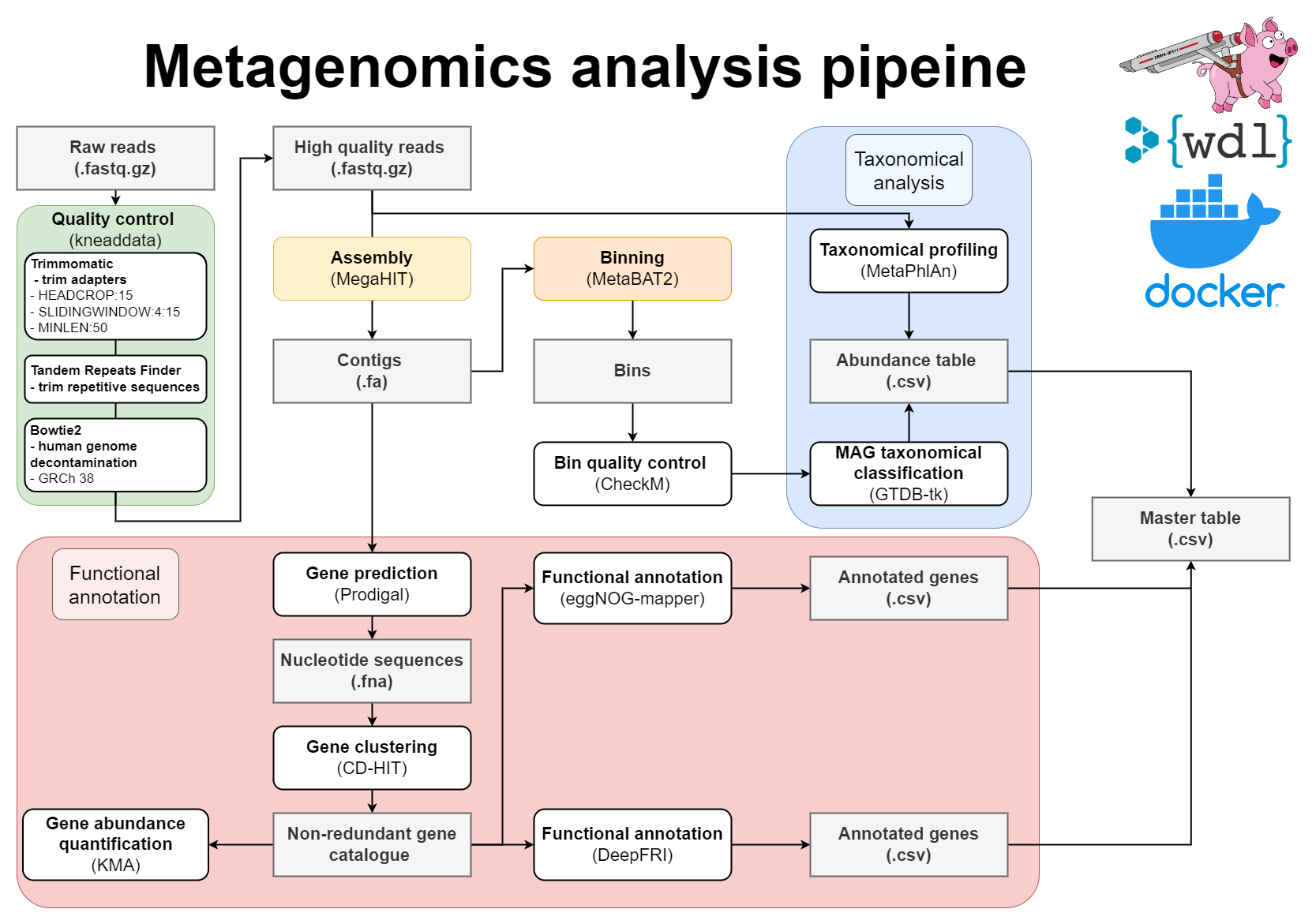
WDL Workflow for metagenome assembly:
 Python script to generate a mapping between non-redundant gene catalogue and MAGS
Python script to generate a mapping between non-redundant gene catalogue and MAGS
The wrapper scripts in Python (located in src) will prepare files and send them to Cromwell. Cromwell executes instructions written in Workflow definition Language (WDL; located in src/wdl). To avoid dependency conflicts Cromwell runs Docker containers with preinstalled software (dockerfiles located in docker).
- Pre-processing of reads with Kneaddata
- Metagenomics assembly with Megahit
- Gene prediction
- Mapping of reads against the contigs
- Metagenome binning using MetaBAT2
- Quality assessment of genome bins
- Taxonomic classifications
- Gene clustering with CD-HIT-EST
- Mapping of reads to gene clusters and computing gene counts
Dockercondafor building the environmentconda env create -f pipeline.yml
- Python
Use the setup_cromwell.py script to download and install it.
- python src/setup_cromwell.py --save_path SAVE_PATH
- Requirements
input_folder- path to directory with paired shotgun sequencing filesbt2_index- path to a directory with a Bowite2 index. In case the folder doesn't contain an index, the user would be proposed to download GRCh38 index used for decontamination of metagenomic samples from human DNA.output_folder- path to a directory where the results will be saved
- Optional arguments
threads- number of threads to use (default: 1)concurrent_jobs- number of concurrent jobs to run (default: 1)
- Output
- quality controlled .fastq.gz files
- assembled contigs in
OUTPUT_DIR/assemble - count table with read counts per sample
# Process the data
python src/qc_and_assemble.py -i input_folder -o OUTPUT_DIR -t 8 -c 3 -bt2_index ./GRCh38_bt2This pipeline will produce a number of directories and files
- assemble; contains assembled contigs
- predictgenes; gene coordinates file (GFF), protein translations and nucleotide sequences in fasta format
- metabat2; binned contigs and a summary report
- CheckM; genome assessment summary report
- gtdbtk; taxonomic classification summary file
- cluster_genes; representative sequences and list of clusters
Python3 script to map non-redundant gene catalogue back to contigs, MAGS and eggNOG annotations
- clustering file - tab-delimited file with cluster ID and gene ID
- Non-redundant gene catalogue (fasta)
- Contig files in fasta
- binned contigs (MAGS) in fasta
- taxonomy files (tsv)
- EggNOG annotation file (tsv)
mapping table (tsv file) that links the non-redundant gene catalogue back to contigs, MAGs and to eggNOG annotations