The docker image of LOCLA is freely accessible on our dockerhub website
LOCLA is a novel genome assembly optimization tool, LOCLA, that iteratively enhances the quality of an assembly by locating sequencing reads on partially assembled scaffolds and thus enable gap filling and further scaffolding. LOCLA utilizes reads of diverse sequencing techniques, e.g., 10x Genomics (10xG) Linked-Reads, PacBio HiFi Reads and Oxford Nanopore Technologies. In our own experiments, we assembled 10x Genomics (10xG) Linked-Reads via Supernova assembler and TGS reads via Canu. Additional tools incorporated in our pipeline include Hybrid Scaffold developed by Bionano Genomics and RagTag.
LOCLA contains one preprocess module and four main modules. The following are the minimum required commands to run each main module separately:
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_GapFilling/ProduceBXList.sh -f draft_bwa_mem_C70M60.sam -a draft.fa -o LCB_GapFilling/
/opt/LCB_GapFilling/Assemble.sh -r /10x_preprocess/nonDupFq/split -o LCB_GapFilling/
/opt/LCB_GapFilling/Fill.sh -a draft.fa -o LCB_GapFilling/
It can be a stand alone module without 10xG Linked-Reads.
/opt/GCB_GapFilling/Fill.sh -a draft.fa -g g-contigs.fa -o GCB_GapFilling/
#rename draft.fa by length first to fit our format
/opt/Rename_byLength.sh -a draft.fa -o LCB_Scaffolding/
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_rename.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_Scaffolding/CandidatePair.sh -f draft_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Assemble.sh -f draft_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -r 10x_preprocess/nonDupFq/split -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Scaffolding.sh -f draft_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_rename.fa -o LCB_Scaffolding/
#rename draft.fa by length first to fit our format
/opt/Rename_byLength.sh -a draft.fa -o GCB_Scaffolding/
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_rename.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/GCB_Scaffolding/CandidatePair.sh -f draft_rename_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_rename_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o GCB_Scaffolding/
/opt/GCB_Scaffolding/Scaffolding.sh -f draft_gcbgf_lbgf_labs_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_rename.fa -g g-contigs.fa -o GCB_Scaffolding/
The modules can be used in any order and iterated several times.The source codes are accessible to all.
For advanced usage see TECHNOTES.md.
In terms of Gap Filling, we recommend applying GCB before LCB to fill in bigger gaps first.
If you only have 10x Genomics linked reads at hand, we propose this pipeline
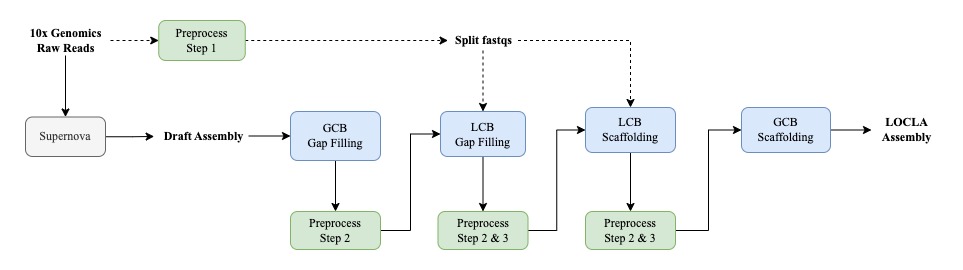
# Categorize fastqs by their barcodes
python /opt/10x_program/step1_preprocessFastq.py -f 10xG_FASTQS --id PROJECTID -o raw_fastq_dir
# GCB Gap-Filling
### Under the premise that only 10x reads are available, we would use the initial draft assembly as g-contigs
/opt/GCB_GapFilling/Fill.sh -a draft.fa -x draft.fa -o GCB_GapFilling/
# LCB Gap-Filling
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_GapFilling/ProduceBXList.sh -f draft_gcbgf_bwa_mem_C70M60.sam -a draft_gcbgf.fa -o LCB_GapFilling/
/opt/LCB_GapFilling/Assemble.sh -r 10x_preprocess/nonDupFq/split/ -o LCB_GapFilling/
/opt/LCB_GapFilling/Fill.sh -a draft_gcbgf.fa -o LCB_GapFilling/
# LCB Scaffolding
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf_lcbgf.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_Scaffolding/CandidatePair.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Assemble.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -r 10x_preprocess/nonDupFq/split -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Scaffolding.s -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_gcbgf_lcbgf.fa -o LCB_Scaffolding/
# GCB Scaffolding
###Under the premise that only 10x reads are available, we would use the latest version of the draft assembly as G-contigs. You could also use draft assemblies from any stage of the pipeline.
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf_lcbgf_lcbs_rename.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/GCB_Scaffolding/CandidatePair.sh -f draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o GCB_Scaffolding/
/opt/GCB_Scaffolding/Scaffolding.sh -f draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_gcbgf_lcbgf_lcbs_rename.fa -g draft_gcbgf_lcbgf_lcbs_rename.fa -o GCB_Scaffolding/
If 10x Genomics Linked-Reads and PacBio or ONT reads (or basically any Third Generation Sequencing long reads) are obtainable, then we suggest this pipeline:

# Categorize fastqs by their barcodes
python /opt/10x_program/step1_preprocessFastq.py -f 10xG_FASTQS --id PROJECTID -o raw_fastq_dir
#GCB Gap-Filling
### If draft.fa is Supernova draft assembly, we recommend you use PacBio/ONT reads or Canu draft assembly as g-contigs.fa
### If draft.fa is Canu draft assembly, we recommend you use Supernova draft assembly or unused PacBio/ONT reads as g-contigs.fa
/opt/GCB_GapFilling/Fill.sh -a draft.fa -g g-contigs.fa -o GCB_GapFilling/
#LCB Gap-Filling
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_GapFilling/ProduceBXList.sh -f draft_gcbgf_bwa_mem_C70M60.sam -a draft_gcbgf.fa -o LCB_GapFilling/
/opt/LCB_GapFilling/Assemble.sh -r 10x_preprocess/nonDupFq/split -o LCB_GapFilling/
/opt/LCB_GapFilling/Fill.sh -a draft_gcbgf.fa -o LCB_GapFilling/
#LCB Scaffolding
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf_lcbgf.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_Scaffolding/CandidatePair.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Assemble.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -r 10x_preprocess/nonDupFq/split -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Scaffolding.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_gcbgf_lcbgf.fa -o LCB_Scaffolding/
#GCB Scaffolding
### If draft.fa is Supernova draft assembly, we recommend you use PacBio/ONT reads or scaffolds from Canu draft assembly that weren’t used in GCB Gap Filling as g-contigs.fa
### If draft.fa is Canu draft assembly, we recommend you use Supernova draft assembly or unused PacBio/ONT reads as g-contigs.fa
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf_lcbgf_lcbs_rename.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/GCB_Scaffolding/CandidatePair.sh -f draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o GCB_Scaffolding/
/opt/GCB_Scaffolding/Scaffolding.sh -f draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_gcbgf_lcbgf_lcbs_rename.fa -g g-contigs.fa -o GCB_Scaffolding/
Pipeline incorporating 10x Genomics linked reads and Bionano Hybrid Scaffold:
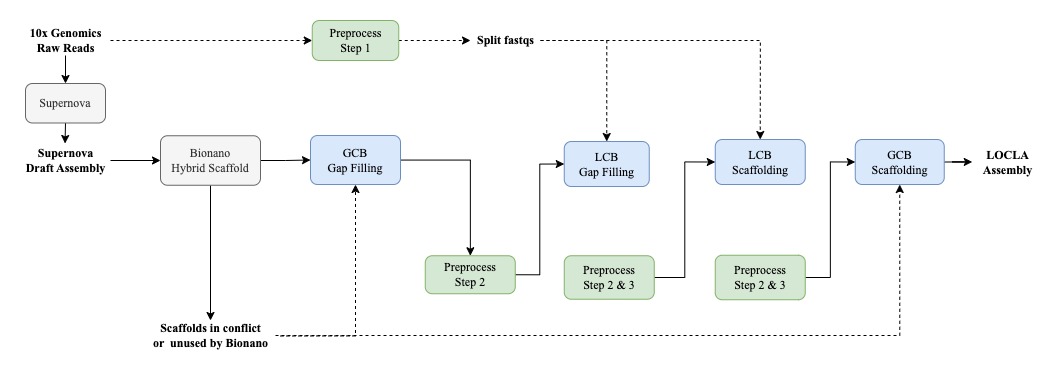
# Categorize fastqs by their barcodes
python /opt/10x_program/step1_preprocessFastq.py -f 10xG_FASTQS --id PROJECTID -o raw_fastq_dir
# GCB Gap-Filling
### We recommend you take scaffolds that are in conflict with and those unused by the Bionano protocol (Hybrid Scaffold only utilizes scaffolds longer than 100kbp) to fill in larger gaps in the Hybrid Scaffold first.
/opt/GCB_GapFilling/Fill.sh -a HybridScaffold.fa -g conflict_unused.fa -o GCB_GapFilling/
# LCB Gap-Filling
### Before running this module, scaffolds in conflict and unused by Bionano should be retrieved; therefore, draft_gcbgf.fa should consist of the Hybrid Scaffold after GCB Gap Filling and conflict_unused.fa in GCB Gap Filling
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_GapFilling/ProduceBXList.sh -f draft_gcbgf_bwa_mem_C70M60.sam -a draft_gcbgf.fa -o LCB_GapFilling/
/opt/LCB_GapFilling/Assemble.sh -r 10x_preprocess/nonDupFq/split -o LCB_GapFilling/
/opt/LCB_GapFilling/Fill.sh -a draft_gcbgf.fa -o LCB_GapFilling/
#LCB Scaffolding
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf_lcbgf.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_Scaffolding/CandidatePair.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Assemble.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -r 10x_preprocess/nonDupFq/split -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Scaffolding.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_gcbgf_lcbgf.fa -o LCB_Scaffolding/
#GCB Scaffolding
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf_lcbgf_lcbs_rename.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/XCB_Scaffolding/CandidatePair.sh -f draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o GCB_Scaffolding/
### G-contigs.fa could be the draft assembly or scaffolds that are in conflict with and those unused by the Bionano protocol
/opt/GCB_Scaffolding/Scaffolding.sh -f draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_gcbgf_lcbgf_lcbs_rename.fa -g g-contigs.fa -o GCB_Scaffolding/
With all resources mentioned above (10x Genomics linked reads, PacBio or ONT reads and Bionano Hybrid Scaffold) available, we suggest this pipeline:
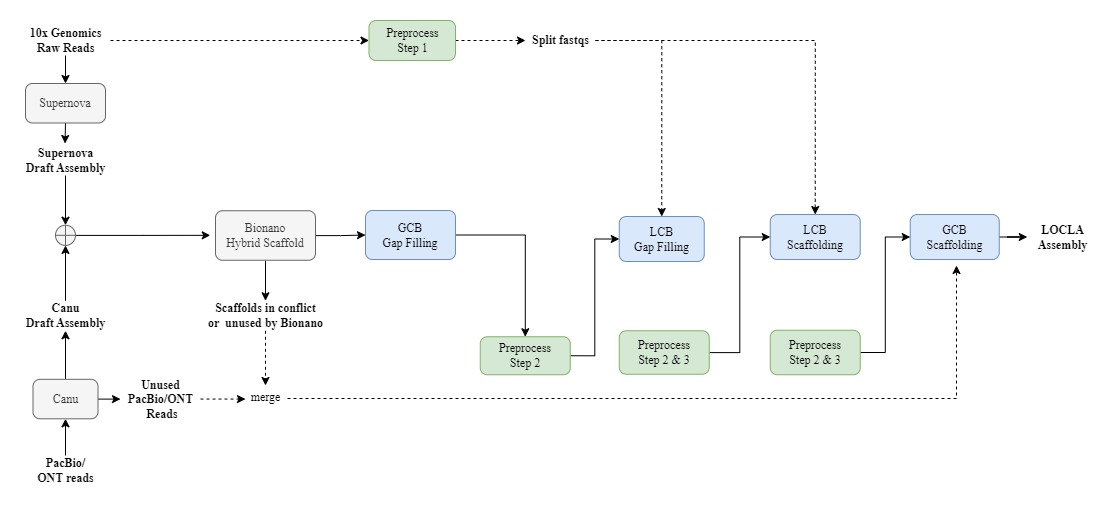
#GCB Gap-Filling
### G-contigs.fa could contain (1) Scaffolds in conflict with Bionano cmap (2) Scaffolds unused by Bionano (3) TGS long reads or draft assembly
We recommend you fill in larger gaps on the Hybrid Scaffold with g-contigs.fa first.
/opt/GCB_GapFilling/Fill.sh -a HybridScaffold.fa -g g-contigs.fa -o GCB_GapFilling/
#LCB Gap-Filling
### Before running this module, scaffolds in conflict and unused by Bionano should be retrieved; therefore, draft_gcbgf.fa should consist of the Hybrid Scaffold after GCB Gap Filling and (1) & (2) aforementioned
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_GapFilling/ProduceBXList.sh -f draft_gcbgf_bwa_mem_C70M60.sam -a draft_gcbgf.fa -o LCB_GapFilling/
/opt/LCB_GapFilling/Assemble.sh -r 10x_preprocess/nonDupFq/split -o LCB_GapFilling/
/opt/LCB_GapFilling/Fill.sh -a draft_gcbgf.fa -o LCB_GapFilling/
#LCB Scaffolding
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf_lcbgf.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/LCB_Scaffolding/CandidatePair.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Assemble.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -r 10x_preprocess/nonDupFq/split -o LCB_Scaffolding/
/opt/LCB_Scaffolding/Scaffolding.sh -f draft_gcbgf_lcbgf_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_gcbgf_lcbgf.fa -o LCB_Scaffolding/
#GCB Scaffolding
python /opt/10x_program/runStep1to3.py -f raw_fastq_dir/ -g draft_gcbgf_lcbgf_lcbs_rename.fa --id PROJECTID -a bwa_mem -o 10x_preprocess/
/opt/GCB_Scaffolding/CandidatePair.sh -f draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafA_ScafB_BXCnt.tsv -p draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafHeadTail_BX_pairSum.tsv -o GCB_Scaffolding/
### Since unused and conflict scaffolds were already added back to the draft assembly. G-contigs.fa could be TGS long reads or scaffolds of draft assembly that weren’t used in GCB Gap Filling
/opt/GCB_Scaffolding/Scaffolding.sh -f draft_gcbgf_lcbgf_lcbs_bwa_mem_C70M60_ScafA_ScafB_BXCnt_rmMultiEnd.tsv -a draft_gcbgf_lcbgf_lcbs_rename.fa -g g-contigs.fa -o GCB_Scaffolding/
You can also run GABOLA without 10x Genomic linked reads. GCB Gap Filling is a module designed for optimizing genome assemblies with PacBio or Nanopore reads. The pipeline suggested below is simplified, different assembling or polishing tools can be performed in between iterations of GCB Gap Filling.
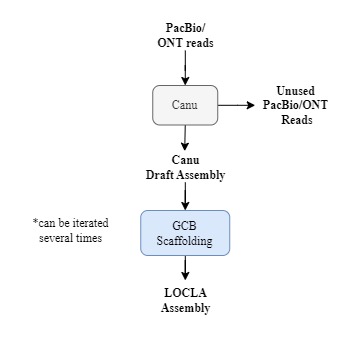
#GCB Gap-Filling
### Unused PacBio/ONT reads as g-contigs.fa
/opt/GCB_GapFilling/Fill.sh -a draft.fa -g g-contigs.fa -o GCB_GapFilling/
#Filter out used PacBio/ONT reads and perform GCB Gap-Filling again with those unused