One important advantage in plant&animal genetic studies is the availability of experimental populations. A Recombinant Inbred Line (RIL) population can be maintained and used over and over again to map all kinds of different traits. Now, RIL population is not confined to bi-parental RIL population. It can be nested association mapping (NAM) population or multiparent advanced generation intercross (MAGIC) population of arbitrary number of founders. I present a method BCFR for constructing bin-map for RILs of arbitrary number of founders. BCFR use identity by state between RILs and their founders in sliding windows, and a pedigree relationship can be optionally used to refine the bin-map, that is, individuals derived from single F1 hybrid can only have two types of alleles.
-
Python (>=3.5.5)
-
Click (>=7.0)
-
Numpy (>=1.13.1)
-
Pandas (>=0.21.0)
-
Matplotlib (>=1.5.0)
Unzip the bcfr package and install
unzip bcfr-master.zip
cd bcfr-master
python setup.py install
- Put the founders in front of all the other samples in VCF file
bgzip input.vcf
tabix input.vcf.gz
bcftools view -S samples.txt input.vcf.gz > output.vcf
- The genotype in VCF file should be imputed and phased(Beagle software)
The software in prepare step is freely available on internet
bcfr converter --v my_genotypes.vcf --d output_dir --n number_of_founder
bcfr birds --n number_of_founder --d output_dir
bcfr birds --n number_of_founder --d output_dir --p pedigree.txt
bcfr plot-hap --r ril_individual_name --d output_dir
Note: the output_dir should be the same directory in all these steps.
pedigree template was offered as foder.txt
bimDic.pkl is the pickled bin map object created by birds command which can be loaded in python:
with open('bimDic.pkl','rb') as f:
bin_size,bin_map=pickle.load(f)
bin_size and bin_map are dictionary with chromosome names as key. bin_size stores the start position and end position of each bin; bin_map stores the genotypes.
the plain bin_size and bim_map file can be made by prepare_geno_for_magicQTL.py
The individual-wise haplotype map data was hpfDic.pkl.
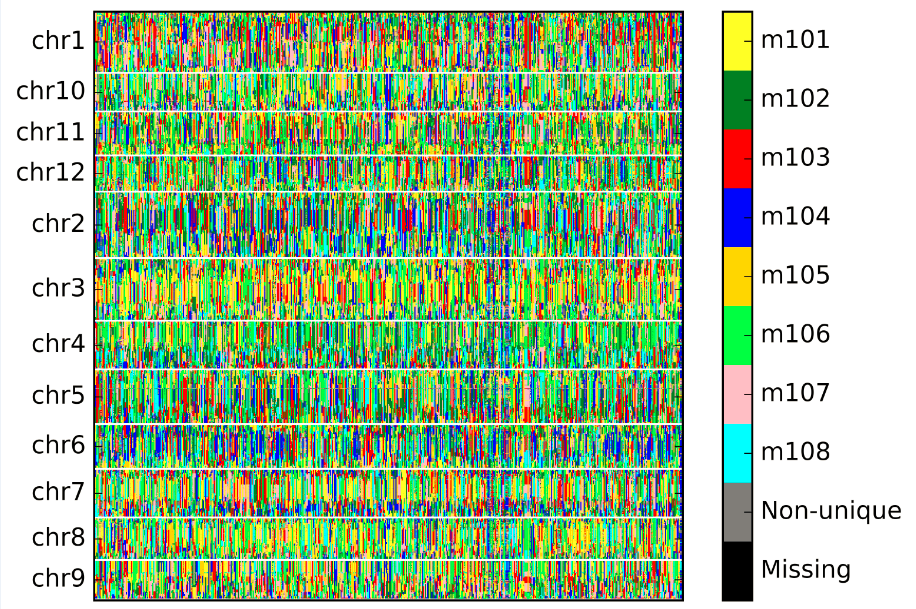
The whole genome bin map chart was output as whole_genome_bin_map.jpg:
The plot-hap command output individual haplotype map chart haplotype_ril_individual_name.jpg:

Calculate bin number,distribution,individual bin number or output a plain file
Start from the output file,make input file for R/qtl, R/MagicQTL
bin2qtl.py
In the output file "CH_for_rqtl.csv", position is in Kb.
prepare_geno_for_magicQTL.py
make_genome_for_magicQTL.r
bcfr --help
bcfr converter --help
bcfr birds --help
Zhongmin Han - National Key Laboratory of Crop Genetic Improvement, Huazhong Agriculture University
Bin-based genome-wide association analyses improve power and resolution in QTL mapping and identify favorable alleles from multiple parents in a 4-way MAGIC rice population. Theoretical and Applied Genetics by Zhongmin Han, Gang Hu, Hua Liu, Famao Liang, Lin Yang, Hu Zhao, Qinghua Zhang, Zhixin Li, Qifa Zhang, Yongzhong Xing
- Lots of memory and fast disk for large projects
-
exgt.py read vcf files from disk write to disk
This script used awk and sed command to extract the genotypes in vcf file. The genotype was splited by homo chromosome. This script perform one row data at each time, so it will not use up a lot memory.
-
df2ss.py read files from disk write to disk (sheets directory)
This script separate coordinates( chrom and position) information and genotype information, Then save data as the format: each chromosome have one coordinate file and one genotype file.
-
flow_across_df.py read from the disk generate variables snkDic and filiDic。
Using sliding windows to calculate the IBS proportion between RILs and parents in a window。
-
ibd_constraint.py read from filiDic.pkl generate variable cstDic
4.1 Convert digits to boolean values, SO we can judge the RIL fragments comes from which parents. If the parents have abs proportion larger than 95%, and differences of the value between parents are smaller than 0.1% , then these parents are treated as the contributor for the RIL fragment and these parents TRUE. (A strict criteria:abs proportion > 99% and number of SNPs in a window > 4).
4.2 The IBD relationships in MAGIC4w pybinXII was finished, but there need more works to let the code commonly used.
-
break_points.py used snkDic and cstDic to generate bksDic
The 5, 6, 7 step handle each individual to make haplotype.
break_points.py judge the breakpoints in each individual. Here we treat the missing haplotype as background gap, so the output data don't have missing value. This treatment makes us easy to adjust the border of each haplotype.
-
opdin_mc.py used xfhDic (the other name of cstDic) and bksDic to generate haprDIc.
Shrink the borders of each haplotype to most upper position that it can reach.
-
maxparsi_mc.py used harprDic to generate mpsDic
Considering all the borders of all the RILs to extend the haplotypes borders with no conflicts. When there are gap with 2 Mb region, the borders between two haplotypes were not extended.
-
divbin.py used xfhDic and mpsDic to generate rwDic,regDic and nbDic.
Delineate bins considering all the borders of all the individuals. Note, the region between a border and the next border was not treated as a bin. if all individuals are missing in a region on the genome , the set all the individual missing(no data on the bin map).
-
retrench.py used regDic and nbDic to generate tulist、bsz and tgd.
Based on the bin map generated by last step impute the bin map: If the upper one row bin and next row bin are all the same except missing values,then merge the two bins.
-
fmb.py used bsz and tgd to generate bimp (bimDic).
After last step imputing, some gaps become smaller, then we can fill the gaps.
-
haplotype_info_indv.py used bsz and bimp to generate tpDic and hpfDic.
Using the constructed bin map to generate haplotypes of each individual. Each individual was a list , this is better expression than 5,6,7 step.
others: double_crossMG.py, grid.py, lover.py is used for treating double-cross problem,they will add dependent on the requirements。 High quality sequencing data don‘t need these three steps.