![]()
fingeRNAt is a software to calculate Structural Interaction Fingerprints in nucleic acids - ligands complexes.
![]()
- Overview
- Installation
- Usage
- Documentation
- Unit test
- Feedback
- Acknowledgments
- How to cite
- License
fingeRNAt is a Python 3.8 script calculating Structural Interaction Fingerprints (SIFs) in complexes of:
| Nucleic acid | Ligand |
|---|---|
| RNA | small molecule ligand |
| RNA | RNA |
| RNA | DNA |
| RNA | protein |
| DNA | small molecule ligand |
| DNA | DNA |
| DNA | RNA |
| DNA | protein |
fingeRNAt calculates different non-covalent interactions between input RNA/DNA structure and ligand and returns long binary string describing if particular interaction occurred between given nucleic acid residue and ligand or not.
fingeRNAt runs under Python 3.5 - 3.8 on Linux, Mac OS and Windows.
Recommended fingeRNAt usage is in conda environment.
- Install conda
Please refer to conda manual and install conda version according to your operating system. Please use Python3 version.
-
Download fingeRNAt repository
Manually - click on the green field
Clone or download, thenDownload ZIPor
Clone it into the desired location [requires git installation]
git clone https://github.com/n-szulc/fingernat.git -
Restore conda environment
conda env create -f fingeRNAt/env/fingeRNAt_env.yml
Required dependencies are:
- Python 3.8
- openbabel 3.1.1
- numpy
- pandas
- matplotlib
- tk
- sphinx
To call fingeRNAt with example inputs:
conda activate fingernat
cd fingeRNAt
python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f SIMPLEwhere:
-r path to RNA/DNA structure; see -> Inputs
-l path to ligands file; see -> Inputs
[-f] optional Structural Interactions Fingerprint (SIFt) type;
available types are: FULL [default], SIMPLE, PBS, XP
see -> SIFs types
[-o] optional path to save output
[-dha] optional Donor-Hydrogen-Acceptor angle calculation when detecting hydrogen bonds;
see -> 1. Hydrogen Bonds
[-vis] optional SIFs results heatmap visualization; see -> Visualization
[-wrapper] optional SIFs results wrapper, see -> Wrappers
available types are: ACUG, PuPy, Counter
[-h] show help message
-r: path to DNA/RNA structure-
supported file types: pdb, mol2
-
only 1 model of DNA/RNA structure
- if there are more models, you have to choose only one (e.g. manually delete remaining models)
-
only DNA/RNA chains
- no water, ions, ligands
We recommend adding hydrogens
- if DNA/RNA structure was obtained from NMR, hydrogens are already there
- if DNA/RNA structure was obtained from XR or cryo-EM, hydrogens can be added using e.g. PyMOL, VMD, Chimera.
-
-
-l: path to small molecule ligands OR DNA/RNA structure- small molecule ligands
- supported file types: sdf, mol2, pdb
- possible multiple poses of ligands in one file
- RNA/DNA structure
- supported file types: pdb, mol2
- possible multiple models of DNA/RNA structure
- only DNA/RNA chains
- no water, ions, ligands
We recommend protonating ligands prior running analysis, e.g. using OpenBabel.
If calculating SIFs
FULLorXPtype, all the missing ligands' hydrogens will be automatically added. - small molecule ligands
Structural Interaction Fingerprint (SIFt) is a binary string, describing existence (1/0) of specified molecular interactions between all target's residues and ligand (Deng et al., 2004).


Fig. 1. Example of SIFt calculated for six non-covalent interactions between HIV-2 Trans-activation response element (TAR) structure (PDB ID: 1AJU) and imatinib.
Available SIFs types [-f]
-
FULLCalculates six non-covalent interactions for each DNA/RNA residue - ligand pair: hydrogen bondings (HB), halogen bondings (HAL), cation - anion (CA), Pi - cation (Pi_Cation), Pi - anion (Pi_anion) & Pi - stacking (Pi_Stacking) interactions; returns six 0/1 values for each residue.
-
SIMPLECalculates distances between each DNA/RNA residue and ligand; returns 1 if the distance is less than declared threshold (default = 4.0 Å), 0 otherwise. Does not take into account distances between hydrogens or hydrogen - heavy atom.
-
PBSDivides each DNA/RNA residue in 3 groups: Phosphate, Base, Sugar and for each group calculates distance to the ligand; returns three 0/1 values for each group within residue - 1 if the distance is less than declared threshold (default = 4.0 Å), 0 otherwise. Does not take into account distances between hydrogens or hydrogen - heavy atom.
NOTE: Only for DNA/RNA with canonical residues.
-
XPCalculates the same six non-covalent interactions for each DNA/RNA residue as
FULL, however it is of no binary type - it calculates total number of each potential interactions occurrence (exception: Pi - interactions) for each RNA/DNA - ligand pair, therefore being an extra precision hologram.NOTE: It returns total number of potential interactions between given residue and ligand pair, e.g. if the residue has one hydrogen bond donor and the ligand has two hydrogen bond acceptors (both fulfilling hydrogen bonds geometrical rules), XP will return
2, despite the fact that one hydrogen bond donor may interact with only one hydrogen bond acceptor.NOTE: It does not calculate total number of potential Pi - interactions as both purine's rings are considered separately. If total numbers of Pi - interactions were calculated, interaction between purine and ligand's aromatic ring would be calculated as two independent interactions, which would not be true.
In case of hydrogen bonds,
XPnot only calculates total number of their potential occurrence in each DNA/RNA - ligand pair, but also assigns each hydrogen bond to strong/moderate/weak type and calculates total number of each of them.
Inspired by PLIP implementation.

Geometric rules:
- |D - A| < 3.9 Å
NOTE: If hydrogens are present in DNA/RNA structure and in ligand, fingeRNAt can be run with flag
-dha, that additionaly calculates Donor-Hydrogen-Acceptor angle used as supplementary criteria in hydrogen bonds detection: 100° < D-H-A angle < 260°
Applies only toFULL/XPSIFt type, asSIMPLE&PBSdo not calculate hydrogen bonds.
(Torshin, Weber, & Harrison, 2002)
In case of XP hologram, there is additional assignment of each hydrogen bond type. Depending on Donor - Acceptor distance, each hydrogen bond can be assigned as strong/moderate/weak.
- 2.2 Å < |D - A| < 2.5 Å: strong
- 2.5 Å < |D - A| < 3.5 Å: moderate, mostly electrostatic
- 3.5 Å < |D - A| < 4.0 Å: weak, electrostatic
(Jeffrey, 1997)

Geometric rules:
- |X - O| < 4.0 Å
- C-X-O angle ~ 165° ± 30°
- X-O-Y angle ~ 120° ± 30°
(Auffinger et al., 2004)

Geometric rule:
- 0.5 Å < |cation - anion| < 5.5 Å
(Barlow and Thornton, 1983)

Geometric rules:
- |cation/anion - aromatic ring center| < 6.0 Å (Gallivan and Dougherty, 1999)
- angle between the ring plane and the line between cation/anion - ring center ~ 90° ± 30°

NOTE: All above interactions' types are considered by fingeRNAt.
Geometric rules:
Common rules for all Pi - stacking interactions' types:
- |rings' centroids| < 5.5 Å (McGaughey, 1998)
- rings' outset < 2.0 Å
For Sandwich & Parallel - displaced:
- angle between the ring planes < 30°
For T - shaped:
- angle between the ring planes ~ 90° ± 30°
All the default thresholds can be changed in code/config.py
Calculated SIFs are saved to tsv files. This is a simple text format similar to csv, except for the data being tab-separated instead of comma-separated (as in csv).
If fingeRNAT was run without optional parameter -o, script will create outputs/ directory in the working directory and save there the output in tsv format. Otherwise fingeRNAt will save outputs in the user-specified location.

Sample extract of output of running python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf

Sample extract of output of running python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f SIMPLE

Sample extract of output of running python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f PBS

Sample extract of output of running python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f XP
Calculated SIFs of all four types can be wrapped, representing them in decreasing resolutions. Multiple wrappers may be passed at once (comma-separated; see -> 'Usage examples'). The results for the SIFs calculations and all passed wrappers are saved to separate tsv files.
There are 3 types of wrappers:
-
ACUGWraps calculated results according to nucleotide, gives information if particular kind of interaction between e.g. any adenine from DNA/RNA and ligand occurred (for SIFt types:
SIMPLE,PBS,FULL) or returns number of possible interactions with all adenines (for SIFt typeXP; see ->XP).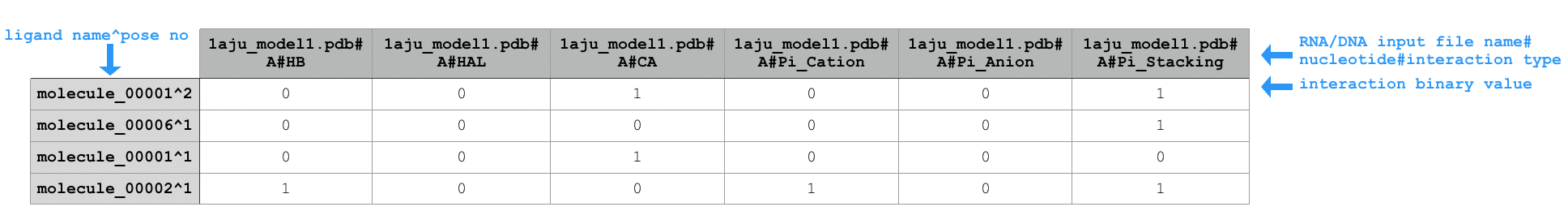
Sample extract of output of running
python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -wrapper ACUG
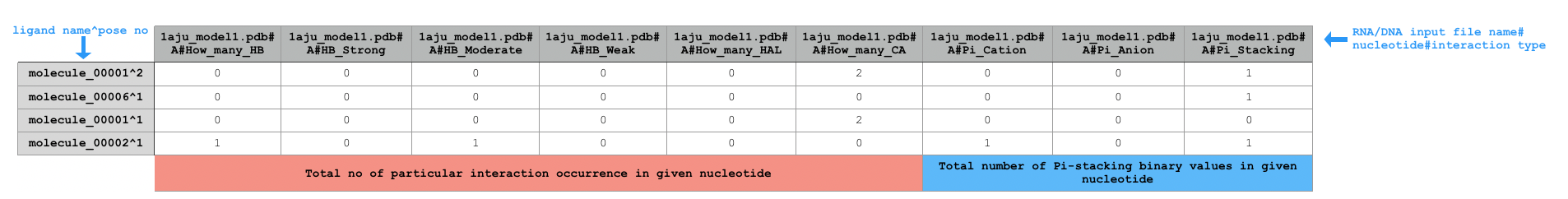
Sample extract of output of running
python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f XP -wrapper ACUG -
PuPyWraps calculated results according to nucleobase type (purine or pyrimidyne), gives information if particular kind of interaction between e.g. any purine from DNA/RNA and ligand occurred (for SIFt types:
SIMPLE,PBS,FULL) or returns number of possible interactions with all purines (for SIFt typeXP; see ->XP).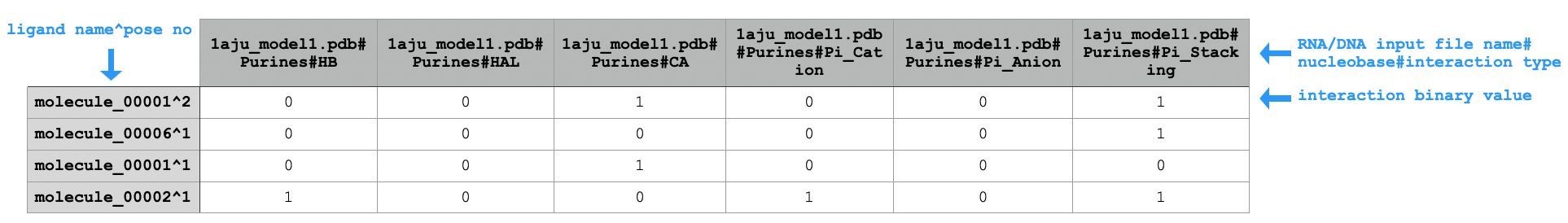
Sample extract of output of running
python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -wrapper PuPy
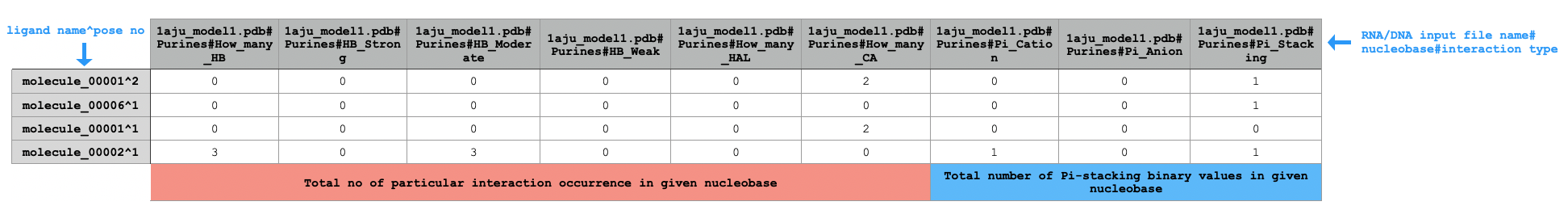
Sample extract of output of running
python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f XP -wrapper PuPy -
CounterCounts total number of given interaction type for any SIFt type. Sums all binary interactions (for SIFt types:
SIMPLE,PBS,FULL) or calculates total number of possible interactions (for SIFt typeXP; see ->XP).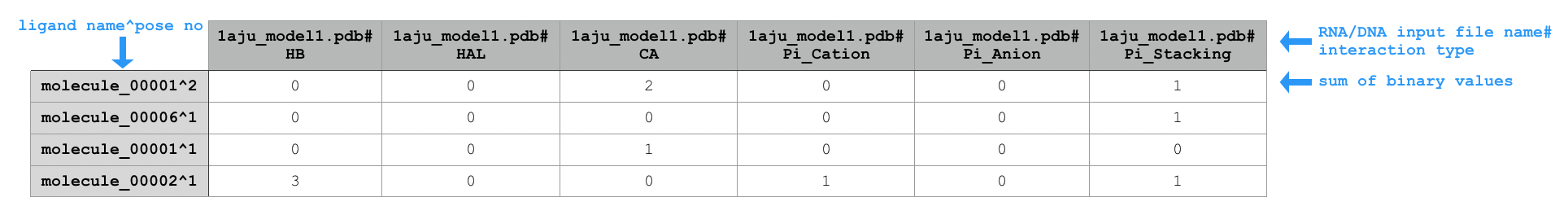
Sample extract of output of running
python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -wrapper Counter

Sample extract of output of running
python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f XP -wrapper Counter






All SIFs outputs can be visualized as heatmap and saved as png files with the same name as tsv output.

Heatmap for SIFt type SIMPLE obtained from running python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f SIMPLE -vis

Heatmap for SIFt type FULL with wrapper ACUG obtained from running python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -vis -wrapper ACUG

Heatmap for SIFt type XP with wrapper PuPy obtained from running python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f XP -vis -wrapper PuPy

Heatmap for SIFt type XP with wrapper Counter obtained from running python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f XP -vis -wrapper Counter
To use Graphical User Interface (GUI), simply run
python code/gui.py
GUI is user-friendly and has all aforementioned functionalities.

-
Calculate SIFt
SIMPLEand save the output in the user-declared location.python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f SIMPLE -o /path/to/my_output -
Calculate SIFt
PBSand save the output with the default name in the current directory together with heatmap.python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f PBS -vis -
Calculate default SIFt
FULLand save it's output and three wrapped outputs with the deafult names in the current directory.python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -wrapper ACUG,PuPy,Counter -
Calculate SIFt
XPconsidering Donor-Hydrogen-Acceptor angle calculation (when detecting hydrogen bonds) and save the output, one wrapped output and two heatmaps in the user-declared location.python code/fingeRNAt.py -r example_inputs/1aju_model1.pdb -l example_inputs/ligands.sdf -f XP -dha -o /path/to/my_output -vis -wrapper ACUG
To generate documentation file using sphinx:
cd docs
make htmlThe documentation will be available from _build/html.
To run a unit test:
cd tests
python fingeRNAt_test.pyWe welcome any feedback, please send an email to Natalia Szulc @n-szulc
Special thanks of gratitude to Masoud Farsani, Pritha Ghosh and Tomasz Wirecki for their invaluable feedback as well as to Prof. Janusz M. Bujnicki and the entire Bujnicki Lab for all the support and project guidelines.
Extensive script testing provided by Zuzanna Mackiewicz has been a great help in developing this tool.
Assistance provided by Open Babel Community was greatly appreciated.
Authors:
Natalia A. Szulc, @n-szulc
Filip Stefaniak, @filipsPL
If you use this software, please cite:
fingeRNAt - a software for analysis of nucleic acids-ligand complexes. Design and applications.
Natalia A. Szulc, Zuzanna Mackiewicz, Janusz M. Bujnicki, Filip Stefaniak [in preparation]
fingeRNAt is licensed under the GNU General Public License v3.0