- How to run Simulations and analyse data
- 0.0 Preparation
- 1. Simulations
- 2. Data Analysis
- 2.1 Files organization
- 2.2 Allele counts: per generation & per replicate
- 2.3 Genome scan: window the genome and calculate pi
- 2.4 Map troughs based on a threshold
- 2.5 Convert diversity data to proportion lost
- 2.6 trough distribuitions depending on recombination landscape (permutation)
- 2.7 trough asymmetry
- 3. Reference to the Figures
- create new folder:
${HOME}/${USER}/fwd_RangeExpansion - create parameter files (for example:
params_1d_c5_e5_l100_d100_f30_m10_g5.sh) and put it in a subfolder:${HOME}/${USER}/fwd_RangeExpansion/param_files - run simulations
chrL: genome length in bpmu: mutation raterho: recombination ratecore: number of demes at the coremaxN: carrying capacity (max num. inds)mig: migration rate in decimal (SLiM way of defining migration)tgrw: time to reach caryying capacityr1:ert1: growth factor at each generationburn: last generation of burn in tree fileaftgen: generation to start expansionend: last expansion generationFndrs: number of founders
# replicates ID numbers
from=1; to=200;
model_prefix="1d_c5_e5_l100_d100"
model_sufix="f20_m10_g5 10"
number_of_samples=10
cd ${HOME}/${USER}/fwd_RangeExpansion
mkdir -p log_sim
sbatch --array=${from}-${to} 01_slurm_run_model_vcf.sh ${model_sufix} ${model_prefix} ${number_of_samples}
This code will:
- make new folder named:
$prefix_$sufix - copy files to model folder:
params_$prefix_$sufix.sh01_run_model_vcf.sh01_1D_range_exp_vcf.slim
- create replicate number subfolders (based on array command from SLURM)
- copy slim file to replicate folder (
01_1D_range_exp_vcf.slim) - creates SLiM log file:
vcf_end_slim_**.output(**: replicate number) - run sims in SLiM:
- output files:
out_p*_gen_2500*_n*.vcf.gz.*in order: (1) population ID; (2) generation; (3) sample size; - samping frequency is defined in SLiM script, depending on the need. Normally based on variables
tg(slim script);tgrw(param file), which define time to reach carrying capacity (from founders to Nmax).
- output files:
file name example: out_p24_gen_25093_n45.vcf
Example for sample size of 45
##fileformat=VCFv4.2
##fileDate=20220822
##source=SLiM
##slimGenomePedigreeIDs=125543612,125543613,125543614,125543615,125543594,125543595,125543618,125543619,125543572,125543573,125543566,125543567,125543592,125543593,125543548,125543549,125543586,125543587,125543558,125543559,125543580,125543581,125543552,125543553,125543542,125543543,125543574,125543575,125543570,125543571,125543554,125543555,125543578,125543579,125543590,125543591,125543560,125543561,125543532,125543533,125543544,125543545,125543568,125543569,125543534,125543535,125543550,125543551,125543596,125543597,125543538,125543539,125543616,125543617,125543536,125543537,125543564,125543565,125543582,125543583,125543540,125543541,125543546,125543547,125543610,125543611,125543584,125543585,125543600,125543601,125543598,125543599,125543556,125543557,125543562,125543563,125543620,125543621,125543608,125543609,125543576,125543577,125543606,125543607,125543602,125543603,125543604,125543605,125543588,125543589
##INFO=<ID=MID,Number=.,Type=Integer,Description="Mutation ID in SLiM">
##INFO=<ID=S,Number=.,Type=Float,Description="Selection Coefficient">
##INFO=<ID=DOM,Number=.,Type=Float,Description="Dominance">
##INFO=<ID=PO,Number=.,Type=Integer,Description="Population of Origin">
##INFO=<ID=GO,Number=.,Type=Integer,Description="Generation of Origin">
##INFO=<ID=MT,Number=.,Type=Integer,Description="Mutation Type">
##INFO=<ID=AC,Number=.,Type=Integer,Description="Allele Count">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Total Depth">
##INFO=<ID=MULTIALLELIC,Number=0,Type=Flag,Description="Multiallelic">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT i0 i1 i2 i3 i4 i5 i6 i7 i8 i9 i10 i11 i12 i13 i14 i15 i16 i17 i18 i19 i20 i21 i22 i23 i24 i25 i26 i27 i28 i29 i30 i31 i32 i33 i34 i35 i36 i37 i38 i39 i40 i41 i42 i43 i44
1 1290 . A T 1000 PASS MID=78192399;S=0;DOM=0;PO=5;GO=12510;MT=1;AC=19;DP=1000 GT 0|1 0|0 0|0 0|0 1|0 0|1 0|0 0|0 0|1 1|0 0|0 0|0 1|0 0|1 0|1 0|1 0|0 0|1 0|0 0|0 1|0 0|1 0|0 0|1 1|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 1|0 0|0 0|0 0|1 1|1 0|0 0|0 0|0 0|0 0|0 0|1
1 2092 . A T 1000 PASS MID=98063402;S=0;DOM=0;PO=1;GO=15690;MT=1;AC=71;DP=1000 GT 1|0 1|1 1|1 1|1 0|1 1|0 1|1 1|1 1|0 0|1 1|1 1|1 0|1 1|0 1|0 1|0 1|1 1|0 1|1 1|1 0|1 1|0 1|1 1|0 0|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 0|1 1|1 1|1 1|0 0|0 1|1 1|1 1|1 1|1 1|1 1|0
1 10065 . A T 1000 PASS MID=61165310;S=0;DOM=0;PO=4;GO=9786;MT=1;AC=87;DP=1000 GT 1|1 1|1 0|1 1|1 1|1 1|1 1|1 1|1 0|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|0 1|1 1|1 1|1 1|1
1 11367 . A T 1000 PASS MID=124563473;S=0;DOM=0;PO=5;GO=19930;MT=1;AC=3;DP=1000 GT 0|0 0|0 1|0 0|0 0|0 0|0 0|0 0|0 1|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|1 0|0 0|0 0|0 0|0
1 12129 . A T 1000 PASS MID=59614940;S=0;DOM=0;PO=2;GO=9539;MT=1;AC=87;DP=1000 GT 1|1 1|1 0|1 1|1 1|1 1|1 1|1 1|1 0|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|1 1|0 1|1 1|1 1|1 1|1blabla back to top ↑
several steps
- samp_edge_demes_1D_10.txt
- 10k_win_coord.csv
- FUNCTIONS_1D_2D_PLOTS.R
step 2
- 02_get_allele_counts_for_pi_calcul.sh
- 02_slurm_get_allele_counts_for_pi_calcul.sh
step 3
- 03_SLURM_Get_Pi_Profile.sh
- 03b_SLURM_conCAT_replicate_files.sh
- 03_FUNCTIONS_Get_Pi_Profile.R
- 03_Get_Pi_Profile.R
step 4
- 04b_slurm_ConCat_trough_files.sh
- 04a_slurm_get_troughs_per_replicate.sh
- 04a_get_troughs_per_replicate.R
- 04_FUNCTIONS_get_troughs_per_replicate.R
These steps below have to happen after steps 1-4, but not in any particular order:
step 5
- 07_convert_time_to_diversity_loss.R
- 07_slurm_convert_time_to_diversity_loss.sh
step 6
- 06_slurm_permutation.sh
- 06_null_dist_troughs.R
- 06_FUNCTIONS_null_dist_troughs.R
- recMap_100k_m**.txt → recombination map file
step 7 (can only be run after step 6)
- 08_asymmetry_in_troughs.R
- 08_slurm_trough_asymmetry.sh
- 08_FUNCTIONS_asymmetry_in_troughs.R
-
samp_edge_demes_1D_**.txt→**: number of samples; no header & space delimited;generation pop id 25002 6 25007 7 25012 8 25017 9 -
**k_win_coord.csv→**: window size in kb, e.g. 10; with header and comma separatedini end wsize mid_win id 18751 28751 10000 23751 4 26251 36251 10000 31251 5 33751 43751 10000 38751 6 41251 51251 10000 46251 7 -
recMap_100k_m**.txt→**: identifier of type of mapiniPOS endPOS rec.rate chunk id 0 299999 0.000000001 1 300000 899999 0.00000001 2 900000 999999 0.0000001 3 1000000 1299999 0.000000001 4 1300000 1899999 0.00000001 5 1900000 1999999 0.0000001 6
cdto model folder- make a sub folder:
sim_files - move all replicate folders into
sim_files - copy all analysis files to model folder
- run scripts in order (see below)
dir=${HOME}/${sim_type}/${model}
cd ${dir}
printf "cd ${dir}\n"
mkdir -p log_allele_count
printf "# $PWD \n"
sim_type="fwd_RangeExpansion";# defines the parent folder for a single spacial dist (1D or 2D)
opt_s02=3;# defines location of sampling file
mprefix="1d_c5_e5_l100_d100"
msufix="f20_m10_g5"
model=${mprefix}_${msufix}
smp_file="10"
script_02=02_slurm_get_allele_counts_for_pi_calcul.sh
printf "sbatch --array=${list_reps} ${script_02} ${sim_type} ${opt_s02} ${model} ${smp_file} \n"
sbatch --array=${list_reps} ${script_02} ${sim_type} ${opt_s02} ${model} ${smp_file}
example command line: sbatch --array=1-200 $script fwd_RangeExpansion 3 $model 10
For a VCF file with 45 individuals:
| POS | size | allele_count | pop | gen |
|---|---|---|---|---|
| 1290 | 90 | 19 | 24 | 25092 |
| 2092 | 90 | 71 | 24 | 25092 |
| 10065 | 90 | 87 | 24 | 25092 |
| 11367 | 90 | 3 | 24 | 25092 |
| 12129 | 90 | 87 | 24 | 25092 |
| 12512 | 90 | 3 | 24 | 25092 |
| 15230 | 90 | 19 | 24 | 25092 |
| 16693 | 90 | 71 | 24 | 25092 |
| 25140 | 90 | 3 | 24 | 25092 |
it's tab separated, headers are not present in file.
dir=${HOME}/${sim_type}/${model}
cd ${dir}
printf "cd ${dir}\n"
mkdir -p log_get_pi
mprefix="1d_c5_e5_l100_d100"
msufix="f20_m10_g5"
model=${mprefix}_${msufix}
founders=20
migration=10
growth=5
samples=10
windows="10k"
active=T
old_demes=F
smp_file="10"
script_s03=03_SLURM_Get_Pi_Profile.sh
printf "${founders} ${migration} ${growth} ${samples} ${active} ${old_demes} ${windows} ${smp_file} \n"
sbatch --array=${list_reps} ${script_s03} ${founders} ${migration} ${growth} ${samples} ${active} ${old_demes} ${windows} ${smp_file}example command line: sbatch --array=1-200 $script 20 10 5 10 T F 10k 10
file name example: genomic_profile_active_demes_f20_m10_g5_r183_resamp_45inds.txt.gz
| gen | cpop | rep | mean_pi_raw | mean_pi_samp | w_id |
|---|---|---|---|---|---|
| 25002 | 6 | 183 | 0.00016 | 0.00016271186440678 | 1 |
| 25002 | 6 | 183 | 5.35555555555556e-05 | 5.44632768361582e-05 | 2 |
| 25002 | 6 | 183 | 0.000177944444444444 | 0.000180960451977401 | 3 |
| 25002 | 6 | 183 | 0.000258444444444444 | 0.000262824858757062 | 4 |
| ... | ... | ... | ... | ... | ... |
| 25002 | 6 | 183 | 0.000145222222222222 | 0.000147683615819209 | 13331 |
| 25002 | 6 | 183 | 0.000194833333333333 | 0.000198135593220339 | 13332 |
| 25002 | 6 | 183 | 0.000120055555555556 | 0.000122090395480226 | 13333 |
| 25002 | 6 | 183 | 0.000112622222222222 | 0.000114531073446328 | 13334 |
| ... | ... | ... | ... | ... | ... |
| 25402 | 86 | 183 | 2.92345679012346e-05 | 2.95630461922597e-05 | 13326 |
| 25402 | 86 | 183 | 2.92345679012346e-05 | 2.95630461922597e-05 | 13327 |
| ... | ... | ... | ... | ... | ... |
| 25402 | 86 | 183 | 0 | 0 | 13333 |
| 25402 | 86 | 183 | 0 | 0 | 13334 |
| 25407 | 87 | 183 | 3.51604938271603e-06 | 3.55555555555553e-06 | 1 |
| ... | ... | ... | ... | ... | ... |
| 25407 | 87 | 183 | 3.33086419753086e-05 | 3.36828963795256e-05 | 6 |
| 25407 | 87 | 183 | 3.33086419753086e-05 | 3.36828963795256e-05 | 7 |
| 25407 | 87 | 183 | 0 | 0 | 8 |
| ... | ... | ... | ... | ... | ... |
| 25497 | 105 | 183 | 1.03111111111111e-05 | 1.04269662921348e-05 | 1 |
| 25497 | 105 | 183 | 0 | 0 | 2 |
| 25497 | 105 | 183 | 0 | 0 | 3 |
| ... | ... | ... | ... | ... | ... |
| 25497 | 105 | 183 | 0 | 0 | 13332 |
| 25497 | 105 | 183 | 0 | 0 | 13333 |
| 25497 | 105 | 183 | 0 | 0 | 13334 |
mkdir -p log_files
level=0.1
samples=10
mprefix="1d_c5_e5_l100_d100"
msufix="f20_m10_g5"
model=${mprefix}_${msufix}
script_s04a=04a_slurm_get_troughs_per_replicate.sh
printf "${model} \n"
printf "${mprefix} ${windows} \n"
mkdir -p genomic_profile_${windows}
sbatch --array=${list_reps_s03} ${script_s04a} ${level} ${mprefix} ${samples} ${windows}example command line: sbatch --array=1-200 $script 0.1 $mprefix 10 10k
file name example: complete_data_lvl_10_f20_m10_g5_45inds_wind_10k_183.txt.gz
| t_id | cpop | rep | gen | n_win | mp_raw | mp_samp | ini | end | mid_tr | size | tot_tr |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6 | 183 | 25002 | 1 | 0 | 0 | 296251 | 306251 | 301251 | 10000 | 499 |
| 2 | 6 | 183 | 25002 | 1 | 6.444e-06 | 6.553e-06 | 341251 | 351251 | 346251 | 10000 | 499 |
| 3 | 6 | 183 | 25002 | 1 | 3.277e-06 | 3.333e-06 | 671251 | 681251 | 676251 | 10000 | 499 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 683 | 29 | 183 | 25117 | 2 | 5.250e-06 | 5.310e-06 | 38658751 | 38676251 | 38667501 | 17500 | 1678 |
| 684 | 29 | 183 | 25117 | 5 | 2.100e-06 | 2.120e-06 | 38876251 | 38916251 | 38896251 | 40000 | 1678 |
| 685 | 29 | 183 | 25117 | 2 | 5.250e-06 | 5.310e-06 | 38936251 | 38953751 | 38945001 | 17500 | 1678 |
| 686 | 29 | 183 | 25117 | 3 | 7.000e-06 | 7.070e-06 | 38958751 | 38983751 | 38971251 | 25000 | 1678 |
file name example: summary_data_lvl_10_f20_m10_g5_45inds_win_10k_183.txt.gz
| cpop | rep | gen | tot_win | mean_size | tot_tr | chr_tr | prop_tr |
|---|---|---|---|---|---|---|---|
| 6 | 183 | 25002 | 601 | 11533.06613 | 499 | 5755000 | 0.05755 |
| 7 | 183 | 25007 | 810 | 11875 | 648 | 7695000 | 0.07695 |
| 8 | 183 | 25012 | 1234 | 12625.82057 | 914 | 11540000 | 0.1154 |
| 9 | 183 | 25017 | 1312 | 12602.6694 | 974 | 12275000 | 0.12275 |
| 10 | 183 | 25022 | 1437 | 13118.2266 | 1015 | 13315000 | 0.13315 |
| 11 | 183 | 25027 | 1805 | 13982.1883 | 1179 | 16485000 | 0.16485 |
| 12 | 183 | 25032 | 2254 | 14723.42733 | 1383 | 20362500 | 0.203625 |
| … | … | … | … | … | … | … | … |
| 100 | 183 | 25472 | 11915 | 110677.9661 | 826 | 91420000 | 0.9142 |
| 101 | 183 | 25477 | 11953 | 110896.312 | 827 | 91711250 | 0.9171125 |
| 102 | 183 | 25482 | 11943 | 109507.1685 | 837 | 91657500 | 0.916575 |
| 103 | 183 | 25487 | 11975 | 112290.6479 | 818 | 91853750 | 0.9185375 |
| 104 | 183 | 25492 | 12021 | 111250 | 829 | 92226250 | 0.9222625 |
| 105 | 183 | 25497 | 12042 | 113990.7407 | 810 | 92332500 | 0.923325 |
# same vars as s04a
samples=10
founders=20
migration=10
growth=5
# vars **different** from s04a
level=10
from=1
to=200
script_s04b=04b_slurm_ConCat_trough_files.sh
printf "sbatch ${script_s04b} ${founders} ${migration} ${growth} ${samples} ${level} ${windows} ${from} ${to} \n"
sbatch ${script_s04b} ${founders} ${migration} ${growth} ${samples} ${level} ${windows} ${from} ${to}
example command line: sbatch --array=1 $script 20 10 5 10 10 10k 1 200
summary_data_lvl_${level}_f${founders}_m${mig}_g${growth}_${inds}inds_win_${janela}.txt.gz
complete_data_lvl_${level}_f${founders}_m${mig}_g${growth}_${inds}inds_win_${janela}.txt.gz
same structure as replicate files, but just combined all replicates into a single file
target_folder="fwd_RangeExpansion";# ------|
main_folder="nope";# |
prefix="1d_c5_l100_d100";# | --> just used to define file location of data
model="f20_m10_g5";# |
sufix="nope";# |
file_location=2;# -------------------------|
from=1
to=200
nsamp=10
slurm_script="07_slurm_convert_time_to_diversity_loss.sh"
cd /storage/homefs/fs19b061/${target_folder}/${prefix}_${model}_${sufix}
printf "${slurm_script} ${main_folder} ${prefix} ${model} ${file_location}"
sbatch --array=1 ${slurm_script} ${main_folder} ${prefix} ${model} ${sufix} ${file_location} ${from} ${to} ${nsamp}example command line: sbatch array=1 $script nope 1d_c5_l100_d100 f20_m10_g5 nope 2 1 200 10
work directory for R script ${HOME}/${USER}/fwd_RangeExpansion/1d_c5_e5_l100_d100_f20_m10_g5/sim_files
diversity_data_**_%s_%s_wsize_10k_10inds_all_reps.txt (**: pi type)
| gen | rep | N | mean | sd | median | qt2 | qt3 | qt4 | qt1 | qt5 | se | ci |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 25002 | 201 | 13334 | 0.000111465 | 8.16E-05 | 9.60E-05 | 5.10E-05 | 9.60E-05 | 0.000154 | 0 | 0.000312 | 7.06E-07 | 1.38E-06 |
| 25007 | 201 | 13334 | 0.000105583 | 8.11E-05 | 9.00E-05 | 4.70E-05 | 9.00E-05 | 0.000147 | 0 | 0.000305 | 7.02E-07 | 1.38E-06 |
| 25012 | 201 | 13334 | 0.000106118 | 8.11E-05 | 9.05E-05 | 4.70E-05 | 9.05E-05 | 0.0001485 | 0 | 0.000306 | 7.03E-07 | 1.38E-06 |
| 25017 | 201 | 13334 | 0.000103887 | 8.16E-05 | 8.75E-05 | 4.55E-05 | 8.75E-05 | 0.0001445 | 0 | 0.0003045 | 7.06E-07 | 1.38E-06 |
| 25022 | 201 | 13334 | 9.67E-05 | 8.04E-05 | 8.05E-05 | 3.75E-05 | 8.05E-05 | 0.000137 | 0 | 0.0002975 | 6.96E-07 | 1.36E-06 |
lvl=10
mod_prefix="1d_c5_l100_d100"
samp=10
winsize="10k"
mod_sufix="f20_m10_g5"
main_dir="${HOME}/${USER}/recMap_fwd_RangeExpansion"
mod_tail_LIST=("recMap_100k_m2" "recMap_100k_m3")
for mod_tail in "${mod_tail_LIST[@]}"; do
echo ${mod_tail}
cd ${main_dir}/${mod_prefix}_${mod_sufix}_${mod_tail}
echo ${PWD}
mkdir -p results_permutation_10k
sbatch --array=1-200%50 06_slurm_permutation.sh ${lvl} ${mod_prefix} ${samp} ${winsize} ${mod_sufix} ${mapType}
done
Resulting file:
proba_rec_rate_in_troughs_lvltroughThresholdAsPercentage_completeModelName_sampleSizeinds_win_windowsizek_rrep.txt
example: proba_rec_rate_in_troughs_lvl10_1d_c5_l100_d100_f20_m10_g5_10inds_win_10k_r198.txt
| gen | cpop | rep | obs.mean.rec | p-val |
|---|---|---|---|---|
| 25002 | 6 | 198 | 1.65E-08 | 0.71 |
| 25007 | 7 | 198 | 1.62E-08 | 1 |
| 25012 | 8 | 198 | 1.68E-08 | 0.04 |
| 25017 | 9 | 198 | 1.64E-08 | 0.57 |
| 25022 | 10 | 198 | 1.66E-08 | 0.51 |
| 25027 | 11 | 198 | 1.67E-08 | 0.36 |
| 25032 | 12 | 198 | 1.65E-08 | 0.6 |
| 25037 | 13 | 198 | 1.67E-08 | 0.33 |
permut_mean_rec_rate_in_troughs_by_case_lvltroughThresholdAsPercentage_completeModelName_sampleSizeinds_win_windowSizek_rrepNumber.txt
| gen | cpop | rep | perm.mean.rec | obs.mean.rec | is.perm.rate.higher | diff |
|---|---|---|---|---|---|---|
| 25003 | 4 | 132 | 1.67645079714128e-08 | 1.65147443650357e-08 | TRUE | 2.49763606377155e-10 |
| 25003 | 4 | 132 | 1.65731280923584e-08 | 1.65147443650357e-08 | TRUE | 5.83837273227164e-11 |
| 25003 | 4 | 132 | 1.66954370533259e-08 | 1.65147443650357e-08 | TRUE | 1.80692688290289e-10 |
| 25003 | 4 | 132 | 1.68642550852116e-08 | 1.65147443650357e-08 | TRUE | 3.49510720175962e-10 |
Name of file where results are stored out.file.name=sprintf("results_permutation_%s/proba_rec_rate_in_troughs_lvl%s_%s_%s_%sinds_win_%s_r%s.txt", winsize, lvl, mod_prefix, mod_sufix, samp, winsize, rep) dist.file.name=sprintf("results_permutation_%s/permut_mean_rec_rate_in_troughs_by_case_lvl%s_%s_%s_%sinds_win_%s_r%s.txt", winsize, lvl, mod_prefix, mod_sufix, samp, winsize, rep)
lvl=10
mod_prefix="1d_c5_l100_d100"
samp=10
winsize="10k"
main_dir="${HOME}/${USER}/recMap_fwd_RangeExpansion"
mod_tail="recMap_100k"
minRecRegionLength=200
# mapType="m4"
# mapType="m6"
mapType="m7"
mod_sufix_LIST=("f20_m10_g5")
for mod_sufix in "${mod_sufix_LIST[@]}"; do
echo ${mod_sufix}
cd ${main_dir}/${mod_prefix}_${mod_sufix}_${mod_tail}_${mapType}
echo ${PWD}
mkdir -p results_asymmetry_${winsize}
sbatch --array=1-200 08_slurm_trough_asymmetry.sh ${lvl} ${mod_prefix} ${samp} ${winsize} ${mod_sufix} ${mapType} ${minRecRegionLength}
doneRscript 08_asymmetry_in_troughs.R 10 1d_c5_l100_d100 10 10k 198 f30_m10_g5 m1 200 &
lvl=10;
mod_prefix=1d_c5_l100_d100;
samp=10;
winsize=10k;
rep=198;
mod_sufix=f30_m10_g5;
mapType=m1;
minRecRegionLength=200;workdir: ${HOME}/recMap_fwd_RangeExpansion/1d_c5_l100_d100_f30_m10_g5_recMap_100k in file: genomic_profile_10k/complete_data_lvl_10_f30_m10_g5_10inds_wind_10k_198.txt.gz
out file NAME example: prop_troughs_in_rec_rate_category_200bp_MinRecRegLen_r198_10lvl_f30_m10_g5_10inds_10kwsize.txt
| gen | cpop | rep | prop.low | prop.med | prop.high |
|---|---|---|---|---|---|
| 25002 | 6 | 198 | 0.308206715 | 0.593076153 | 0.098694386 |
| 25007 | 7 | 198 | 0.312335798 | 0.587299648 | 0.100344189 |
| 25012 | 8 | 198 | 0.316670454 | 0.587802015 | 0.095508597 |
| … | … | … | … | … | … |
| 25487 | 103 | 198 | 0.300391549 | 0.599650677 | 0.099955375 |
| 25492 | 104 | 198 | 0.300516609 | 0.59982661 | 0.099654407 |
| 25497 | 105 | 198 | 0.301468522 | 0.598972342 | 0.099556775 |
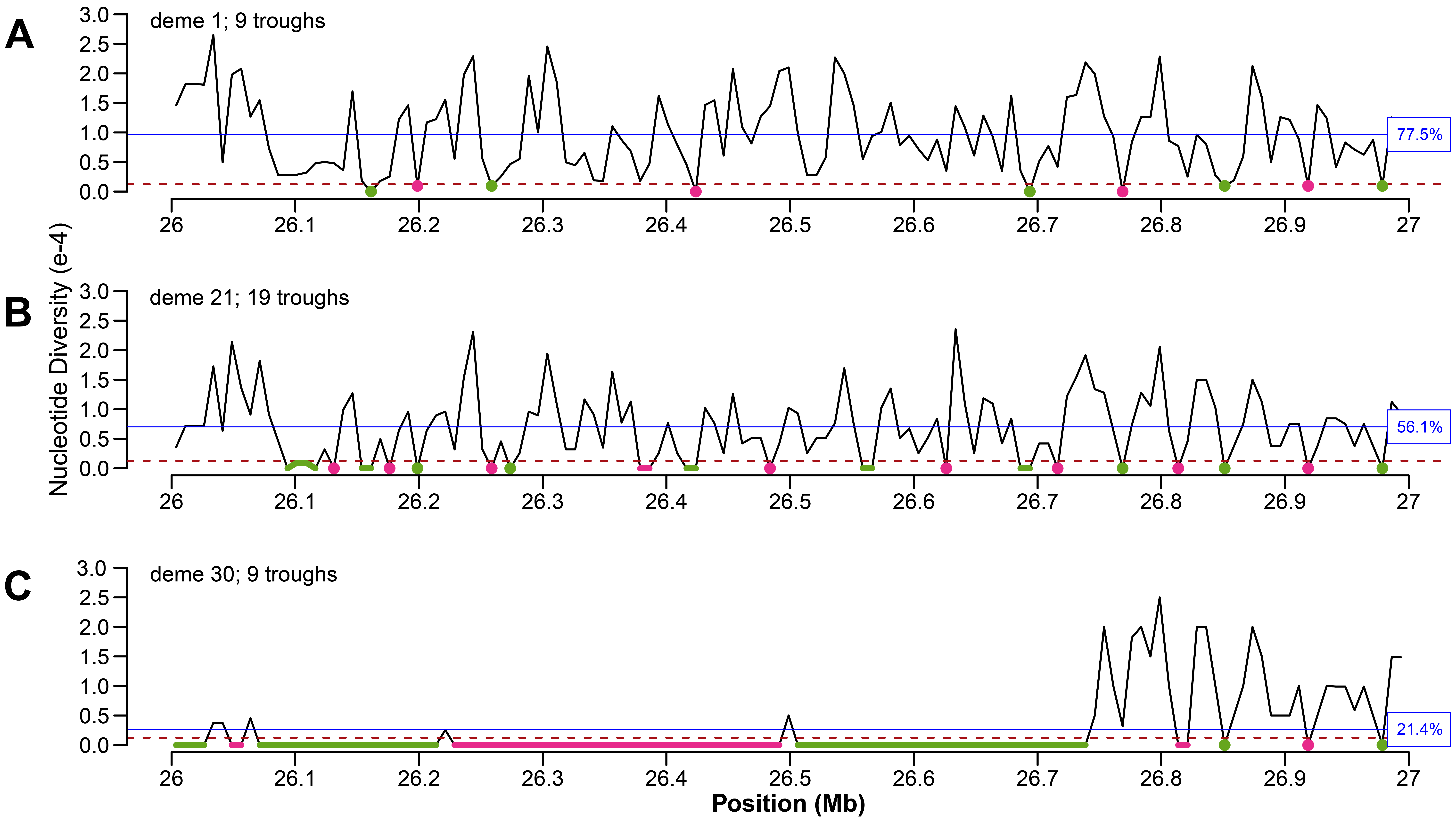
Figure 1 in the main text
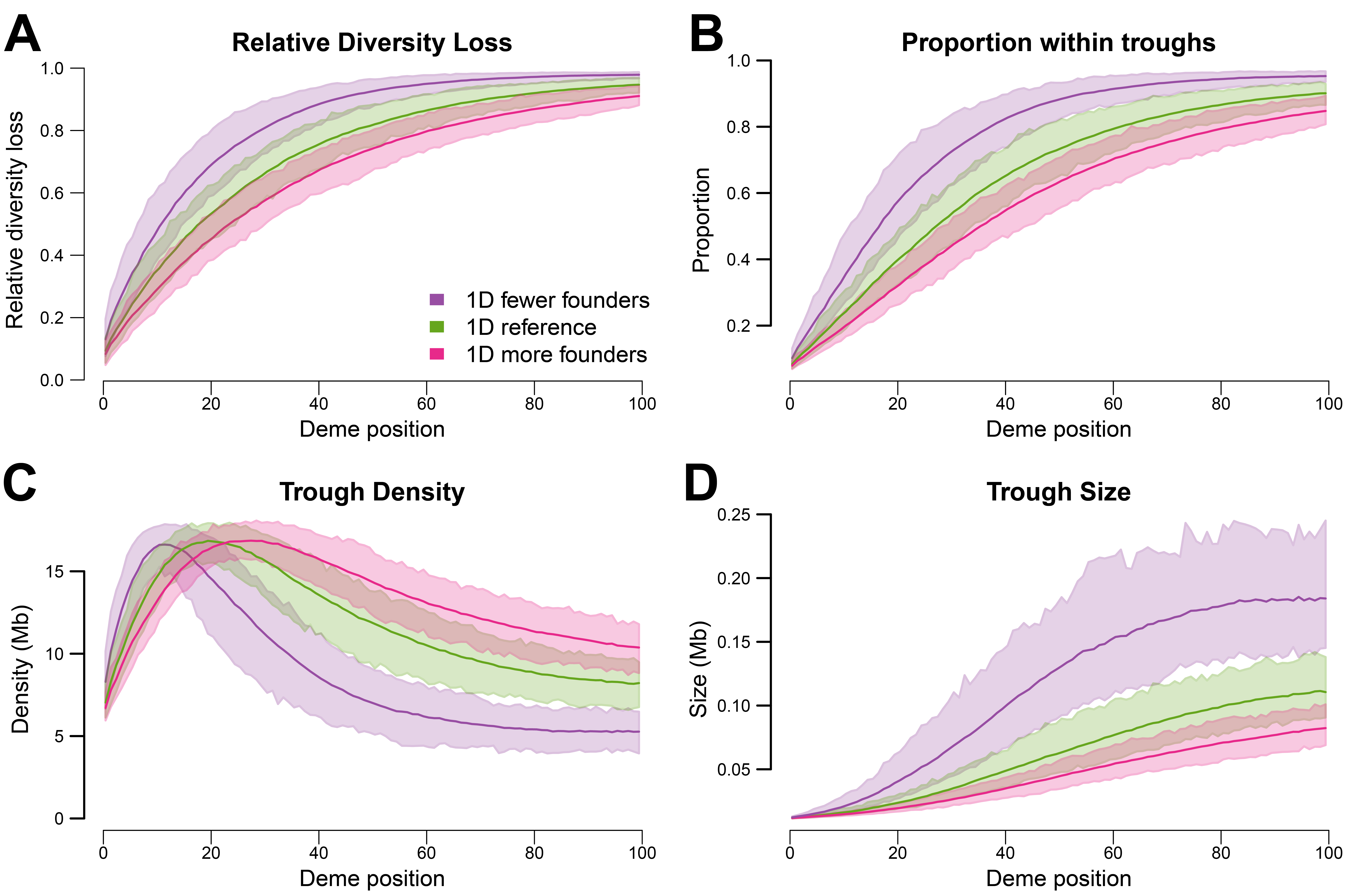
Figure 2 in the main text

Figure 3 in the main text
Steps used:
- Allele counts (s02)
- window the genome and calculate pi (s03)
- find troughs (s04).
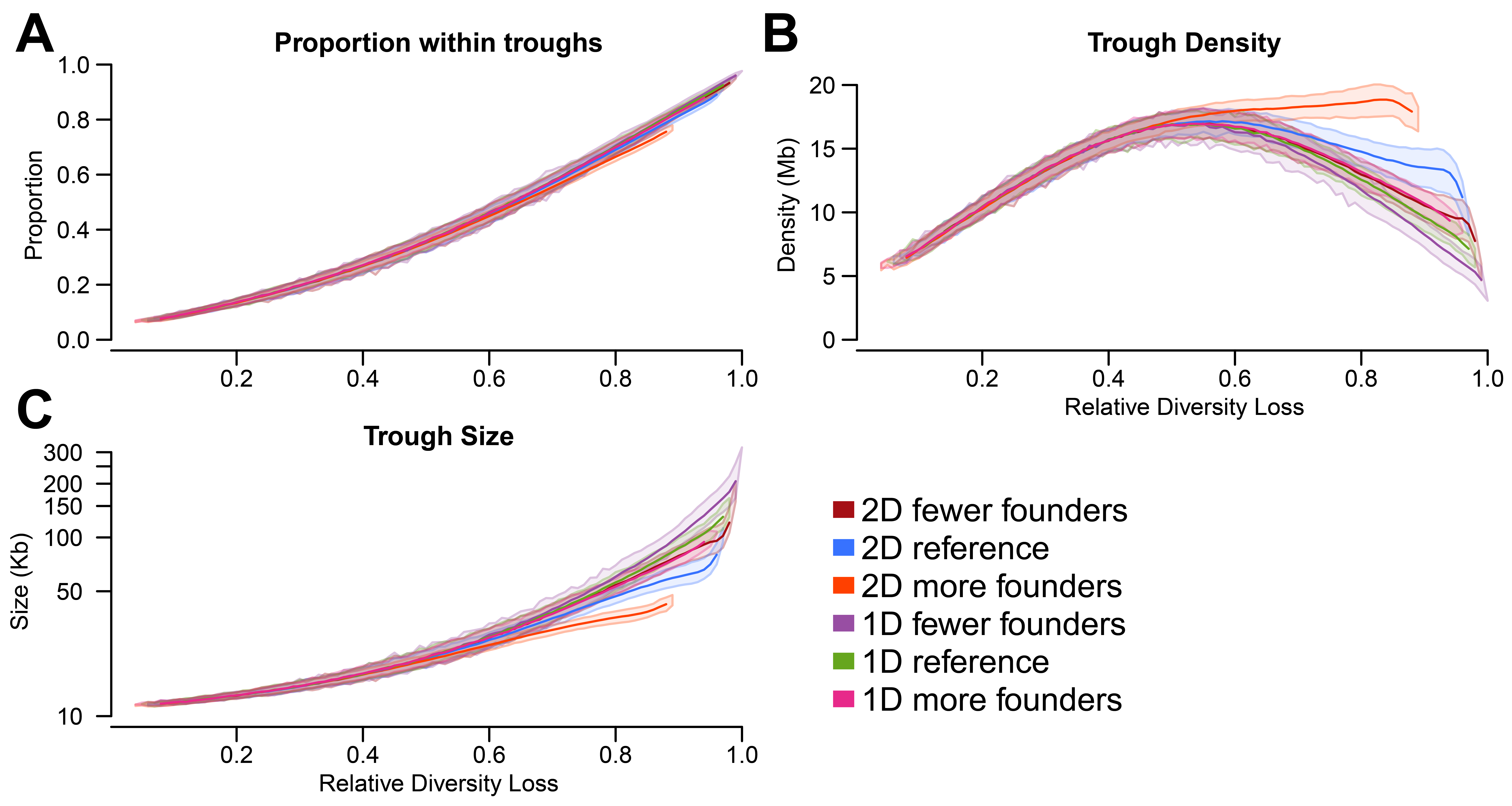
Figure 4 in the main text
Steps used:
- Proportion of diversity loss (s07) back to top ↑
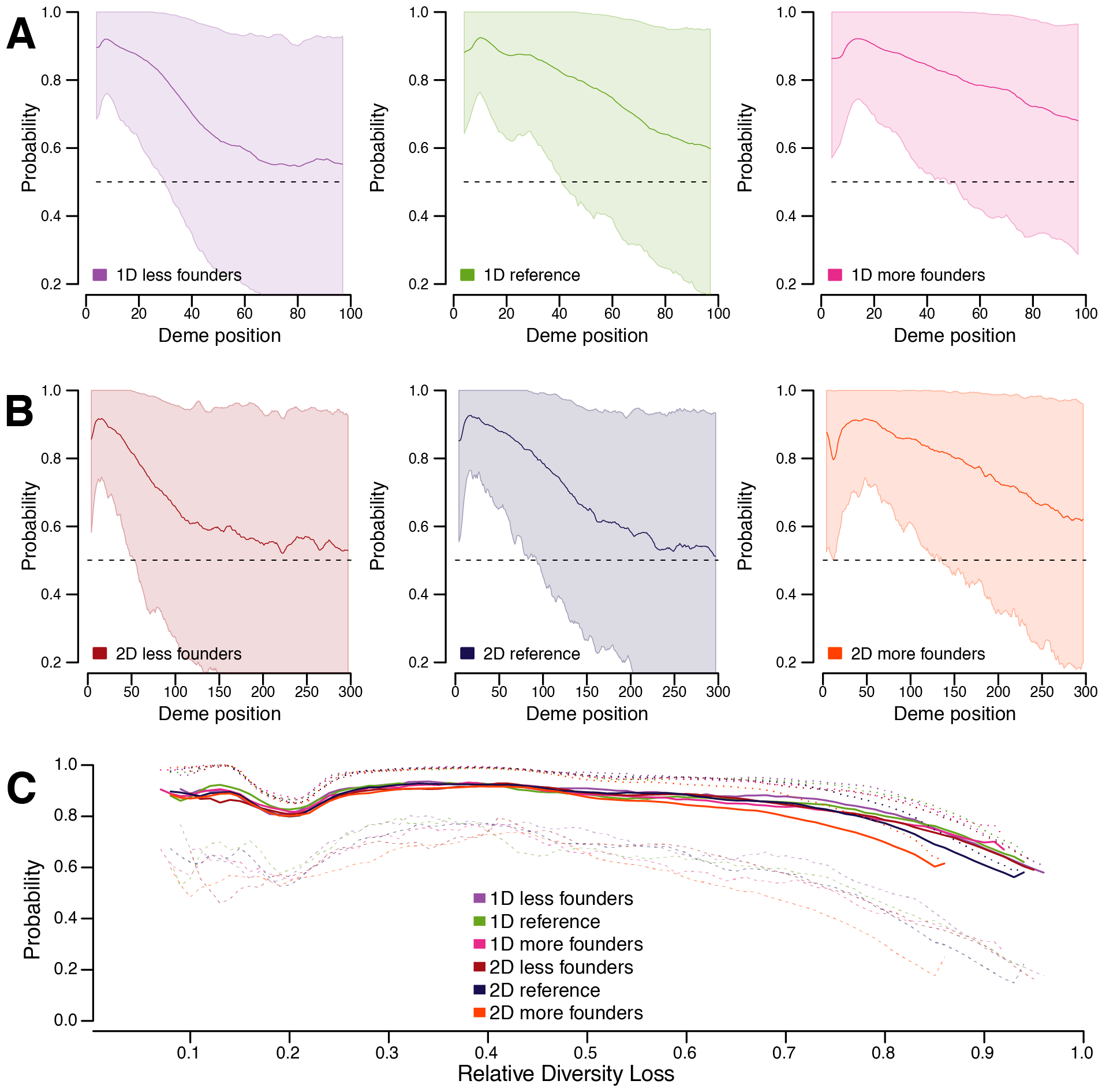
Figure 5 in the main text
Steps used:
- permutation analysis (s06)
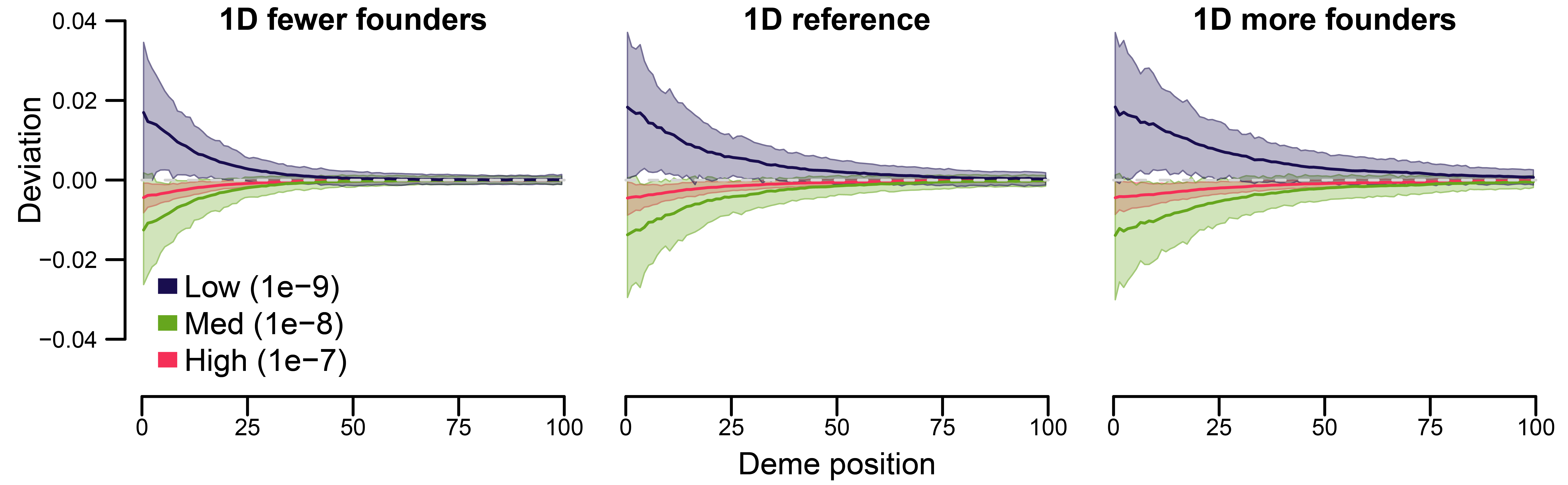
Figure 6 in the main text
Steps used:
- assymetry in troughs (s08)