This is a package for evaluating Graph Embedding prediction methodologies. GraphGuest works on any kind of heterogeneous undirected graphs with exact 2 types of nodes and 1 type of edge. It was developed in the context of drug repurposing, as a part of the paper "Towards a more inductive world for drug repurposing approaches". From now on, we will refer to the Type 1 and 2 nodes as Drugs and Proteins, respectively, hence the evaluated graph would be a Drug Target Interaction (DTI) network.
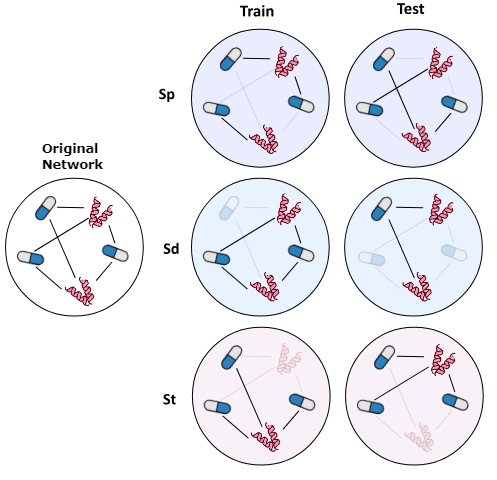
GraphGuest allows to split any chosen network into train/test folds following several criteria and configurations:
- Random: There are no constraints imposed, DTIs are distributed across train and test randomly.
- Sp: Related to pairs. Any drug or protein may appear both in the train and test set, but interactions cannot be duplicated in the two sets.
- Sd: Related to drug nodes. Drug nodes are not duplicated in the train and test set, i.e., a drug node evaluated during training does not appear in the test set.
- St: Related to targets. Protein nodes are not duplicated in the train and test set, therefore each protein seen during training does not appear in the test set.
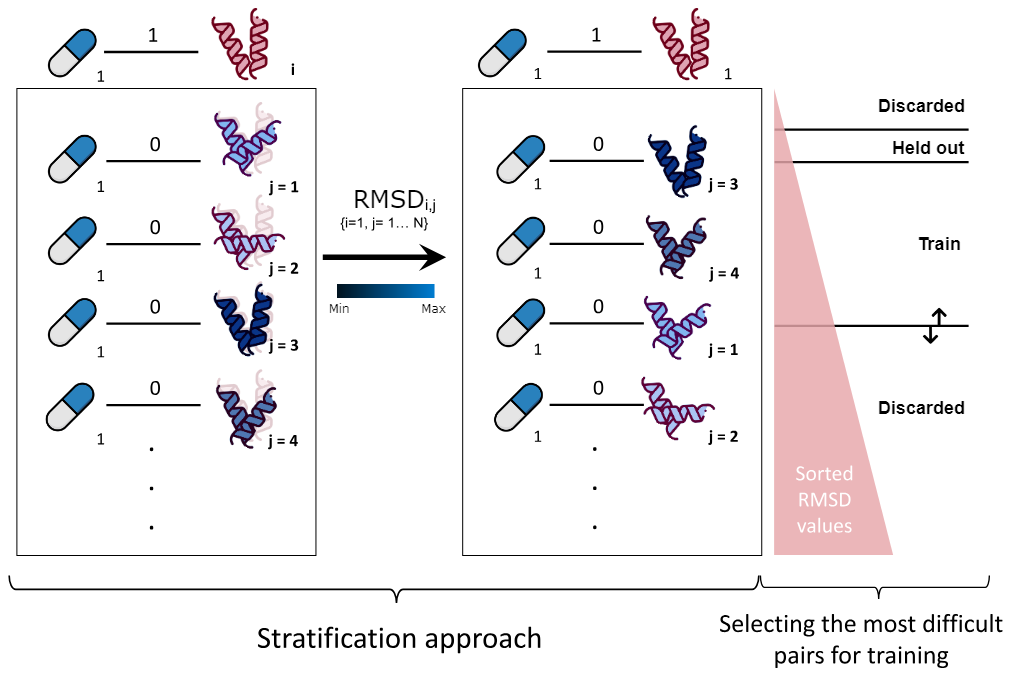
Generally DTI networks are highly sparse, i.e., there is a high number of negative interactions compared to the positive ones. Hence, including all negative edges is not feasible, and would bias the model towards negative predictions. Accordingly, usually a balanced dataset is built by selecting all the positive interactions and subsampling the same number (negative to positive ratio of 1) of negatives randomly. In the presented work, we showed that random subsampling can oversimplify the prediction task, as it is likely that the model is not evaluated on hard-to-classify negative samples. Also, this subsampling methodology lacks of biological meaning. Hence, we propose to rank negative interactions based on a structural-based metric (RMSD of the distance between atoms of two protein structures) to find hard-to-classify samples and increase accuracy and robustness of the drug repurposing model.
In this line, GraphGuest allows to use a matrix of distances/scores between every Protein as an alternative to random subsampling. If this matrix is provided, for each positive DTI, the negative DTI will be formed by the same drug and the protein that better maximizes (or minimizes) the distance/score with respect to the original protein from the positive DTI.
Here now we describe the functionalities and parameters of the GraphGuest GUEST class:
- DTIs: Interaction list in the form of a pandas matrix with the columns "Drug" and "Protein" as the type 1 and 2 nodes.
- mode: The already introduced split criteria: random, Sp, Sd or St (default: Sp).
- subsampling: Whether all interactions are chosen to build the dataset or subsampling is preferred instead (default: True).
- n_seeds: Number of times the dataset will be built, varying the seed, hence yielding different splits (default: 5).
- negative_to_positive_ratio: How many negatives DTI will be subsampled respect to the positives ones (default: 1).
After installing GraphGuest with pip (pip install graphguest), the required packages can be imported:
from graphguest import GUEST
import pandas as pd
import pickle
Then, load the DTI dataset. It must be a pandas matrix containing the columns "Drug" and "Protein". An example of the Yamanishi's NR network is located in the test folder (nr_dti.txt).
DTIs = pd.read_csv("tests/nr_dti.txt", sep='\t', header=None)
DTIs.columns = ['Drug', 'Protein']
Load the GUEST object, specifying the DTI dataset, the mode you want the dataset to fulfill, as well as subsampling options and number of seeds.
ggnr = GUEST(DTIs, mode = "Sp", subsampling = True, n_seeds = 5)
You can optionally pass a Protein column's score matrix as an argument. This matrix is built by computing the Root Mean Square Devitation (RMSD) between every pair of proteins (atomic distances) in the evaluated dataset. As a result, negative subsampling will shift from random selection to a rank-based approach. For each Drug-Target Interaction (DTI), negative DTIs will be selected according to their rank and a predefined threshold. Here the discarded RMSD values are <=2.5 Å, the held out are >2.5 & <=5 Å and >5 & <=6 Å (see RMSD Figure).
# Example matrix containing random values
ggnr.apply_rank(RMSD_threshold = (2.5, 5, 6), fpath = 'tests/rmsd_nr.pkl')
Now, generate the splits according to the specified options. GraphGuest can generate n folds fulfilling split criteria in a Cross-Validation fashion, or following a Train/Val/Test configuration.
ggnr.generate_splits_cv(foldnum=10) #(Cross-Validation)
ggnr.generate_splits_tvt() #(Train-Validation-Test)
Finally, retrieve the results. If RMSD option has been applied, the held-out fold will be returned (See RMSD Figure). Also, a node embedding dictionary can be passed as an argument to generate the node embedding datasets according to the generated split distribution.
## Load the node embedding dictionary
# We randomly generate (2,) and (3,) shaped arrays for drugs and proteins, respectively
with open('tests/node_emb_nr.pkl', 'rb') as handle:
node_emb = pickle.load(handle)
## Retrieve the results
# Default behavior
seed_cv_list = ggnr.retrieve_results()
# Passing a node embedding dictionary
seed_cv_list, seed_cv_ne = ggnr.retrieve_results(node_emb)
# When score/RMSD matrix is provided (apply_rank method called)
seed_cv_list, final_fold = ggnr.retrieve_results()
# If previous options (rank matrix + node_emb dict) are combined
seed_cv_list, final_fold, seed_cv_ne = ggnr.retrieve_results(node_emb)
You can verify that your splits fulfill the mode requirements after they have been generated. Note that if the apply_rank method has been called, the split mode argument will be ignored due to inconsistencies between the rank and the split constraints (Sp, Sd or St constraints may not be possible to fulfill if a rank-based constraint has been imposed as well.)
# Default behavior
ggnr.test_splits()
# Verbose can be set to True if more information is desired. (verbose's default: False)
ggnr.test_splits(verbose=True)
# You can also visualize the final distribution of DTIs by setting distr to True (distr's default: False)
ggnr.test_splits(distr=True)