Neil F. Thompson, Eric C. Anderson, Anthony Clemento, Matthew Campbell, Devon E. Pearse, James Hearsey, Andrew P. Kinziger, John Carlos Garza
Last Updated: 2020-05-15
This repository includes code, data, and some intermediate results to reproduce the results in Thompson et al. (“A complex phenotype in salmon controlled by a simple change in migratory timing”). You can get the whole thing by cloning or downloading the repo from https://github.com/eriqande/thompson-et-al-2020-chinook-salmon-migration-timing.
If you are viewing this as the README of a GitHub repository, note that you can read it in a somewhat friendlier format (i.e., with a table of contents, etc.) on GitHub pages at: https://eriqande.github.io/thompson-et-al-2020-chinook-salmon-migration-timing/
For the whole-genome resequencing data, the production of BAMs and VCFs from the original FASTQ files follows a standard bwa/GATK pipeline as described in the supplemental methods. Here we focus on analyses of the products of that work. The various steps are described in a series of RMarkdown documents with a fair bit of explanation/description of the procedures. Analyses requiring the use of cluster computing resources are described and documented, but will not be run automatically by evaluating the RMarkdown document. Implementing them yourself will require modification for the vagaries of your own high performance computing system and preferred mode of parallelization, etc. Running some of the RMarkdown documents requires a proper shell. These were initially developed and run on a Mac. They have also been tested and confirmed to run on a server running CentOs 7.
Rmarkdown documents in the 000 series were developed primarily by Eric C. Anderson. Rmarkdown documents in the 100 series were developed primarily by Neil F. Thompson. Rmarkdown documents in the 200 series were developed primarily by Eric C. Anderson and deal with further analyses done to respond to several reviewer queries.
In most cases, products from cluster computing are placed in this
repository within the stored_results directory. This means that all
the RMarkdown documents can be evaluated successfully without first
running the cluster-based analyses, except for
002-allele-frequencies.Rmd, for which the products from the
cluster-based analyses require too much space to store in this
repository. Fortunately, none of the downstream analyses depend upon the
outputs from 002-allele-frequencies.Rmd.
All the RMarkdown documents can be evaluated en masse (except for
002-allele-frequencies.Rmd) by sourcing the R script
render-numbered-Rmds.R. On a fairly old mac laptop the run times for
each are as follows. Note that this does not include the time required
to create the stored results on the cluster,
etc.
| Rmarkdown document | Run time |
|---|---|
| 001-align-coho-genome.Rmd | 1.7 secs |
| 002-allele-frequencies.Rmd (after running initial results on cluster) | 3.3 mins |
| 003-extract-johnson-creek-variants.Rmd | 8.7 mins |
| 004-prepare-haplotypes.Rmd | 11.2 mins |
| 005-annotating-variants-near-greb1l.Rmd | 5.9 mins |
| 006-haplo-raster-plots.Rmd | 1.2 mins |
| 007-arg-inference-and-plotting.Rmd | 4.9 mins |
| 008-read-depths-and-duplications.Rmd | 41.2 secs |
| 009-rosa-dna-distance-trees.Rmd | 2.3 mins |
| 010-salmon-river-carcasses.Rmd | 48.2 secs |
| 011-recombinant-frequencies.Rmd | 1.5 mins |
| 012-coalescent-modeling-of-recombinants.Rmd | 3.5 mins |
| 100-RoSA-figure1-map.Rmd | 16.4 secs |
| 101-Klamath-estuary-GSI-rubias.Rmd | 18.0 secs |
| 102-Klamath-estuary-ANOVA-sampling-date-RoSA.Rmd | 11.0 secs |
| 102.1-Klamath-estuary-partial-correlation.Rmd | 1.6 secs |
| 103-Klamath-estuary-GonadSI-mixed-model-analysis.Rmd | 3.3 secs |
| 103.1-Klamath-estuary-GonadSI-power-analysis.Rmd | 1.4 mins |
| 104-Klamath-estuary-nonwaterfraction-adiposity-mixed-model-analysis.Rmd | 2.1 secs |
| 104.1-Klamath-estuary-nonwaterfraction-power-analysis.Rmd | 1.7 mins |
| 105-Klamath-estuary-figure4.Rmd | 7.7 secs |
| 106-TrinityRiver-ANOVA-etc.Rmd | 9.5 secs |
| 107-RoSA-population-genetics-survey.Rmd | 1.6 secs |
| 108-Klamath-basin-early-RoSA-haplotype-abundance-commercial-fishery-model-tableS10.Rmd | 1.2 secs |
| 201-01-prep-bams-to-seek-inversions.Rmd | 0.16 secs |
| 201-02-seek-inversions.Rmd | 3.7 mins |
| 201-03-seek-inversions-with-gridss.Rmd | 4.0 secs |
| 202-01-genomewide-allele-freqs-from-the-bams.Rmd | 4.7 secs |
| 203-01-pca-for-gwas-covariate.Rmd | 4.7 secs |
| 203-02-gwas-with-angsd.Rmd | 4.4 mins |
| 204-01-assess-genotyping-error-rate-with-whoa.Rmd | 1.2 mins |
| 204-02-assess-genotype-error-from-subsampled-hi-read-depth-samples.Rmd | 2.0 mins |
| 204-03-simulate-imputation-and-phasing-error-for-trees.Rmd | 6.7 mins |
Below is a description of the needed dependencies. A script to install
all of them on one of our test clusters is at
000-00-prepare-dependencies.sh, but it will likely need some tweaking
on your system.
Download the necessary Chinook and coho genomes and put them in a new
directory called genome in this repository. Index them with samtools
faidx.
mkdir genome
cd genome
# coho genome
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/021/735/GCF_002021735.1_Okis_V1/GCF_002021735.1_Okis_V1_genomic.fna.gz
mv GCF_002021735.1_Okis_V1_genomic.fna.gz Okis_V1_genomic.fna.gz
gunzip Okis_V1_genomic.fna.gz
samtools faidx Okis_V1_genomic.fna
# chinook genome
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/872/995/GCF_002872995.1_Otsh_v1.0/GCF_002872995.1_Otsh_v1.0_genomic.fna.gz
mv GCF_002872995.1_Otsh_v1.0_genomic.fna.gz Otsh_V1_genomic.fna.gz
gunzip Otsh_V1_genomic.fna.gz
samtools faidx Otsh_V1_genomic.fna
# chinook genome GFF
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/872/995/GCF_002872995.1_Otsh_v1.0/GCF_002872995.1_Otsh_v1.0_genomic.gff.gz
# Narum et al. version of Chinook genome from the Johnson Creek fish
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/vertebrate_other/Oncorhynchus_tshawytscha/all_assembly_versions/GCA_002831465.1_CHI06/GCA_002831465.1_CHI06_genomic.fna.gz
gunzip GCA_002831465.1_CHI06_genomic.fna.gz
samtools faidx GCA_002831465.1_CHI06_genomic.fna
cd ../Make a directory in the top level of the repository called geo-spatial
and then put some things in there:
-
Get CalWater2.2.1 by going to https://drive.google.com/file/d/13WqsA3jK1C0kW5zaznEwDkg3wBc-cBco/view and then choosing the Google Drive download button to download a whole zip file that expands into a directory named
GISlayers_calw221_shp. Move or symlink that whole directory insidegeo-spatial. These are watershed boundaries. -
Get California stream line data from http://data-cdfw.opendata.arcgis.com/datasets/29c40f65341749b3aa26d3f0e09502b9_4.zip. Unzip that directory into one called
California_Streamsand move or symlink that directory intogeo-spatial. -
Download some things from Natural Earth Data:
- Natural Earth II with Shaded Relief, Water, and Drainages raster
https://www.naturalearthdata.com/http//www.naturalearthdata.com/download/10m/raster/NE2_HR_LC_SR_W_DR.zip.
Put or symlink the resulting directory,
NE2_HR_LC_SR_W_DRintogeo-spatial. - 10m-cultural-vectors, Admin 1 – States, Provinces, Download
boundary lines:
https://www.naturalearthdata.com/http//www.naturalearthdata.com/download/10m/cultural/ne_10m_admin_1_states_provinces_lines.zip.
Put or symlink the resulting directory,
ne_10m_admin_1_states_provinces_linesintogeo-spatial. - Finally, get the coastlines:
https://www.naturalearthdata.com/http//www.naturalearthdata.com/download/10m/physical/ne_10m_coastline.zip,
and put the resulting folder,
ne_10m_coastlineintogeo-spatial.
- Natural Earth II with Shaded Relief, Water, and Drainages raster
https://www.naturalearthdata.com/http//www.naturalearthdata.com/download/10m/raster/NE2_HR_LC_SR_W_DR.zip.
Put or symlink the resulting directory,
The following programs must be installed and available in the PATH. Versions used on Eric’s laptop appear in parentheses.
-
angsd(Version: 0.920, using htslib: 1.6) -
bwa(Version: 0.7.16a-r1181) -
samtools(Version: 1.3, using htslib 1.3) -
bcftools(Version: 1.9, using htslib 1.9) -
bgzip,tabix(Version 1.6) These are found within the htslib library. -
vcftools(Version v0.1.12b) -
bedtools(Version v2.27.1) -
PHASE(Version 2.1.1) -
lastz(Version 1.04) This was obtained and compiled thus:wget http://www.bx.psu.edu/~rsharris/lastz/lastz-1.04.03.tar.gz gunzip lastz-1.04.03.tar.gz tar -xvf lastz-1.04.03.tar cd lastz-distrib-1.04.03/ make make install -
maf2fasta,single_cov2(FrommultizVersion 012109) Obtain and compile like this:wget http://www.bx.psu.edu/miller_lab/dist/multiz-tba.012109.tar.gz gunzip multiz-tba.012109.tar.gz tar -xvf multiz-tba.012109.tar cd multiz-tba.012109/ make
The following Java-based programs must be downloaded, and the paths to
their associated Jar files must be listed appropriately in the file
script/java-jar-paths.R:
The distributed version of script/java-jar-paths.R reflects the
placement of Jar files on Eric’s system:
# central place in repository to store paths for Java Jar files we use
BEAGLE=/Users/eriq/Documents/others_code/BEAGLE_4.1/beagle.27Jan18.7e1.jar
RentPlus=/Users/eriq/Documents/others_code/RentPlus/RentPlus.jarThis should be updated to reflect where you have stored the jar files on your system.
One more Java program—snpEff—is required. The installation procedure for this is slightly different, owing to it needing to store data bases in the directory where the Jar file resides (apparently).
So,
- Download
snpEffThe link provided is for Version 4.3 with a release date of 24 November 2017. THis is the version we used. - Expand that zip archive and move the resulting directory into
the top level of this repository. When you are done, you should be able tolsthe Jar file like this:
ls snpEff_v4_3t_core/snpEff/snpEff.jar Packages must be downloaded from CRAN, BioConductor, and GitHub. The
following code, which is in R/install_packages_etc.R, will download
and install the necessary packages:
# just R code to install the packages needed for running the notebooks
# get the packages needed from CRAN
install.packages(
c(
"ape",
"broom",
"callr",
"car",
"cowplot",
"ggsn",
"ggspatial",
"kableExtra",
"knitr",
"lubridate",
"maps",
"mapproj",
"nlme",
"pander",
"parallel",
"phangorn",
"plotly",
"ppcor",
"raster",
"remotes",
"rmarkdown",
"rubias",
"sessioninfo",
"sf",
"sjPlot",
"tidyverse",
"vcfR",
"viridis",
"whoa",
"zoo"
),
repos = "http://cran.rstudio.com"
)
# Note that, on the cluster, since I don't have admin access,
# I needed to get the sysadmin to get rgdal installed and to
# install libudunits2.so in order to complete the install of:
# units, rgdal, sf, rosm, lwgeom, ggsn, ggspatial
# get the packages needed from BioConductor
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager", repos = "http://cran.rstudio.com")
}
BiocManager::install("ggtree")
BiocManager::install("treeio")
# Get package ecaRbioinf at https://github.com/eriqande/ecaRbioinf
# This is a package of useful functions by Eric C. Anderson. We
# used commit `6972defd`. Get it like this:
remotes::install_github("eriqande/ecaRbioinf", ref = "6972defd")The following RMarkdown documents should be evaluated in order. The
script render-numbered-Rmds.R will do that (except for
002-allele-frequencies.Rmd) when run in the top level of this
repository. Some RMarkdown documents rely on outputs from previous ones.
Some of these RMarkdown documents include calls to Unix utilities, so
might not run on non-Unix or non-Linux architectures.
Outputs (figures, tables, R data objects, etc) from each RMarkdown
document are written to the outputs/XXX directories. Intermediate
files written during evaluation of each RMarkdown document are written
to the intermediates/XXX directories. To facilitate working between
the cluster and a desktop/laptop, some outputs are written to the
stored_results/XXX directories which are version controlled and
included in this repo.
Thumbnails of the figures and tables generated by each RMarkdown document appear below. Additionally, at the top of each section is a link to the compiled (HTML-version) of the RMarkdown document on GitHub Pages.
(Compiled RMarkdown HTML document on GitHub Pages: 001-align-coho-genome.html)
The products of this analysis go into creation of some of the trees
(009) and also in the designation of the alleles as ancestral or not
in some of the haplotype rasters (006).
(Compiled RMarkdown HTML document on GitHub Pages: 002-allele-frequencies.html)
Calculation of allele frequency differences between early- and late-running forms across the genome, and around GREB1L, and the generation of plots.

(Compiled RMarkdown HTML document on GitHub Pages: 003-extract-johnson-creek-variants.html)
Using the previously assembled Chinook salmon genome from Narum et al. (2018) to ascertain variation from that Upper Columbia River summer/spring-run fish around the GREB1L region. The products of this analysis go into some later trees and haplotype rasters.
(Compiled RMarkdown HTML document on GitHub Pages: 004-prepare-haplotypes.html)
Using BEAGLE to impute genotypes and infer haplotypes in 5 Mb around GREB1L. The outputs of this analysis are used for a number of downstream analyses.
(Compiled RMarkdown HTML document on GitHub Pages: 005-annotating-variants-near-greb1l.html)
Using snpEff to annotate variants and then analyzing a few of them. This provides data that goes into Table S1 (“Two near-perfectly associated, non-synonymous variants in the GREB1L gene”), and is also used later for the haplotype raster plots.

(Compiled RMarkdown HTML document on GitHub Pages: 006-haplo-raster-plots.html)
Compiling some auxiliary information (gene/exon inclusion, etc.) and then making haplotype raster plots using functions from the ‘ecaRbioinf’ package.
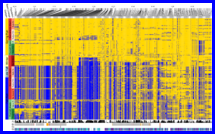
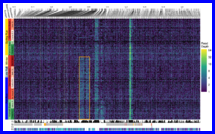
(Compiled RMarkdown HTML document on GitHub Pages: 007-arg-inference-and-plotting.html)
Using RENT+ (Mirzaei and Wu 2016) to infer the ancestral recombination graph in the RoSA and plot it with a tree/heatmap, using ggtree (Yu et al. 2017).

(Compiled RMarkdown HTML document on GitHub Pages: 008-read-depths-and-duplications.html)
Using bedtools to compute read depths from the BAMs and then demonstrate that the apparent duplications are highly associated with run type.
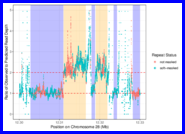

(Compiled RMarkdown HTML document on GitHub Pages: 009-rosa-dna-distance-trees.html)
Making distance trees between haplotypes in the RoSA region, and including coho salmon.
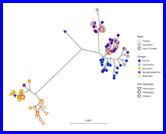

(Compiled RMarkdown HTML document on GitHub Pages: 010-salmon-river-carcasses.html)
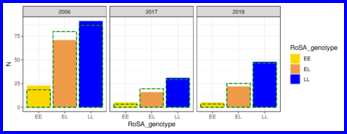
Genotype frequency, spatial and temporal analyses of RoSA genotypes of post-spawn Salmon River carcasses. Data from here also go into Table S7 (“Summary of number of Salmon River Chinook salmon carcasses and their RoSA genotypes”).



(Compiled RMarkdown HTML document on GitHub Pages: 011-recombinant-frequencies.html)
Determining frequencies of recombinant haplotypes between the RoSA region and the imperfectly associated SNPs reported in Prince et al. (2017). Data from this RMarkdown document go into Table S9 (“Frequencies of haplotypes recombinant/non-recombinant between the distal and RoSA region in collections from California and Oregon”).

(Compiled RMarkdown HTML document on GitHub Pages: 012-coalescent-modeling-of-recombinants.html)
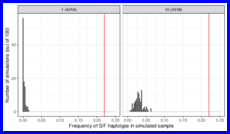
Performing discrete-time, coalescent-with-recombination simulations to determine whether we expect to see such a high recombinant frequency in the Klamath River — Iron Gate Hatchery collection if spring-fall introgression only occurred after human modifications to the watershed.

(Compiled RMarkdown HTML document on GitHub Pages: 100-RoSA-figure1-map-using-sf.html)
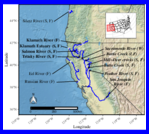
Making the map for Figure 1.
(Compiled RMarkdown HTML document on GitHub Pages: 101-Klamath-estuary-GSI-rubias.html)
Genetic stock identification of Chinook salmon caught in the Klamath River estuary.
(Compiled RMarkdown HTML document on GitHub Pages: 102-Klamath-estuary-ANOVA-sampling-date-RoSA.html)

(Compiled RMarkdown HTML document on GitHub Pages: 103-Klamath-estuary-GonadSI-mixed-model-analysis.html)

(Compiled RMarkdown HTML document on GitHub Pages: 103.1-Klamath-estuary-GonadSI-power-analysis.html)
(Compiled RMarkdown HTML document on GitHub Pages: 104-Klamath-estuary-nonwaterfraction-adiposity-mixed-model-analysis.html)
(Compiled RMarkdown HTML document on GitHub Pages: 104.1-Klamath-estuary-nonwaterfraction-power-analysis.html)
(Compiled RMarkdown HTML document on GitHub Pages: 105-Klamath-estuary-figure4.html)

(Compiled RMarkdown HTML document on GitHub Pages: 106-TrinityRiver-ANOVA-etc.html)


(Compiled RMarkdown HTML document on GitHub Pages: 107-RoSA-population-genetics-survey.html)

(Compiled RMarkdown HTML document on GitHub Pages: 108-Klamath-basin-early-RoSA-haplotype-abundance-commercial-fishery-model-tableS10.html)

(Compiled RMarkdown HTML document on GitHub Pages: 201-01-prep-bams-to-seek-inversions.html)
(Compiled RMarkdown HTML document on GitHub Pages: 201-02-seek-inversions.html)
(Compiled RMarkdown HTML document on GitHub Pages: 201-03-seek-inversions-with-gridss.html)
(Compiled RMarkdown HTML document on GitHub Pages: 202-01-genomewide-allele-freqs-from-the-bams.html)
(Compiled RMarkdown HTML document on GitHub Pages: 203-01-pca-for-gwas-covariate.html)
(Compiled RMarkdown HTML document on GitHub Pages: 203-02-gwas-with-angsd.html)
(Compiled RMarkdown HTML document on GitHub Pages: 204-01-assess-genotyping-error-rate-with-whoa.html)
(Compiled RMarkdown HTML document on GitHub Pages: 204-02-assess-genotype-error-from-subsampled-hi-read-depth-samples.html)
(Compiled RMarkdown HTML document on GitHub Pages: 204-03-simulate-imputation-and-phasing-error-for-trees.html)
Mirzaei, Sajad, and Yufeng Wu. 2016. “RENT+: An Improved Method for Inferring Local Genealogical Trees from Haplotypes with Recombination.” Bioinformatics 33 (7). Oxford University Press: 1021–30.
Narum, Shawn R., Alex Di Genova, Steven J. Micheletti, and Alejandro Maass. 2018. “Genomic Variation Underlying Complex Life-History Traits Revealed by Genome Sequencing in Chinook Salmon.” Proceedings of the Royal Society B: Biological Sciences 285 (1883): 20180935. https://doi.org/10.1098/rspb.2018.0935.
Prince, Daniel J., Sean M. O’Rourke, Tasha Q. Thompson, Omar A. Ali, Hannah S. Lyman, Ismail K. Saglam, Thomas J. Hotaling, Adrian P. Spidle, and Michael R. Miller. 2017. “The Evolutionary Basis of Premature Migration in Pacific Salmon Highlights the Utility of Genomics for Informing Conservation.” Science Advances 3 (8): e1603198. https://doi.org/10.1126/sciadv.1603198.
Yu, Guangchuang, David K. Smith, Huachen Zhu, Yi Guan, and Tommy Tsan-Yuk Lam. 2017. “Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data.” Methods in Ecology and Evolution 8 (1): 28–36. https://doi.org/10.1111/2041-210X.12628.