

Performs Fusion-gene discovery from junction reads identified by STAR during alignment. See citation of STAR-Fusion below.
- Nextflow : for common installation procedures see the IARC-nf repository.
- STAR-Fusion
In addition, STAR-Fusion requires a CTAT bundle with reference genome and annotations.
A conda receipe, and docker and singularity containers are available with all the tools needed to run the pipeline (see "Usage")
| Type | Description |
|---|---|
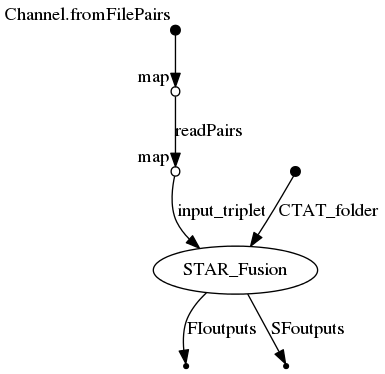
| input_folder | Folder containing fastq files and STAR junction files |
Specify the test files location
| Name | Example value | Description |
|---|---|---|
| --CTAT_folder | . | Folder with STAR-Fusion bundle (CTAT) |
| Name | Default value | Description |
|---|---|---|
| --input_file | NULL | Input file (comma-separated) with 4 columns: SM(sample name), pair1 (path to fastq pair 1), pair2 (path to fastq pair 2), and junction (path to junction file) |
| --output_folder | results_fusion | Output folder |
| --fastq_ext | fq.gz | Extension of fastq files |
| --suffix1 | _1 | Suffix of 1st element of fastq files pair |
| --suffix2 | _2 | Suffix of 2nd element of fastq files pair |
| --junction_suffix | Chimeric.SJ.out.junction | Suffix of STAR chimeric junction files |
| --starfusion_path | /usr/local/src/STAR-Fusion/STAR-Fusion | Path to STAR-fusion executable; note that the default is set to the location in the singularity container |
| --cpu | 2 | Number of cpu used by bwa mem and sambamba |
| --mem | 2 | Size of memory used for mapping (in GB) |
Note: using the input_file mode allows to specify multiple fastq files for a given sample, that are merged during the alignment phase.
Flags are special parameters without value.
| Name | Description |
|---|---|
| --junctions | Option to use STAR junction files already generated |
| --help | Display help |
Note: when the --junctions option is not used, the junction column of the input file is ignored.
nextflow run iarcbioinfo/RNAseq-fusion-nf -r v1.1 -profile singularity --input_folder input --CTAT_folder CTAT --output_folder output
To run the pipeline without singularity just remove "-profile singularity"; you can also directly download a singularity image at https://data.broadinstitute.org/Trinity/CTAT_SINGULARITY/STAR-Fusion/ using the command singularity pull https://data.broadinstitute.org/Trinity/CTAT_SINGULARITY/STAR-Fusion/star-fusion.v1.9.0.simg. Alternatively, one can run the pipeline using a docker container (-profile docker) the conda receipe containing all required dependencies (-profile conda).
| Type | Description |
|---|---|
| output | Folder with fusion genes file |
| Name | Description | |
|---|---|---|
| Nicolas Alcala | alcalan@iarc.fr | Developer to contact for support |
Haas, B. J., Dobin, A., Li, B., Stransky, N., Pochet, N., & Regev, A. (2019). Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome biology, 20(1), 213.